
Начиная с 80-х гг. прошлого века все случаи стремительно развивающейся гемолитической анемии и тромбоцитопении во второй половине беременности или в раннем послеродовом периоде у исходно здоровых женщин расценивались как особый вид тяжелой преэклампсии (ПЭ). В 1982 году L.Weinstein предложил использовать акроним HELLP, включающий Hemolysis – гемолиз, Elevated Liver Enzimes – повышение уровня аспарагинаминотрансферазы (АСТ), аланинаминотрансферазы (АЛТ) и Low Platelets – тромбоцитопению [1]. При этом признаки ПЭ у таких пациенток даже в настоящее время выявляются только в 80%. Кроме того, логически вытекающая из предположения о главной роли беременности в развитии данного состояния, попытка остановить прогрессирование гемолиза и тромбоцитопении путем родоразрешения оказалась далеко не всегда эффективной: практически у трети пациентов явления анемии, тромбоцитопении, поражения печени и почек не только не регрессировали, но и продолжали нарастать.
Кроме того, по данным В. Sibai (1995), у женщин, у которых HELLP-синдром развился или прогрессирует после родоразрешения, выше риск развития острой почечной и дыхательной недостаточности [2–4]. Практически сразу после описания HELLP-синдрома появились описания неполных или парциальных его форм, в которых отсутствует какой-либо компонент, либо на первый план выходит внепеченочное поражение. Тяжелую ПЭ и полный/парциальный HELLP-синдром объединяет тот факт, что все эти состояния представлены симптомокомплексом тромботической микроангиопатии (ТМА).
Общие понятия об акушерских ТМА
ТМА – это клинико-морфологический синдром, проявляющийся микроангиопатической гемолитической анемией (МАГА) и тромбоцитопенией, которые развиваются вследствие окклюзии сосудов микроциркуляторного русла различных органов тромбами, содержащими агрегированные тромбоциты и фибрин. Основным признаком механического микроангиопатического гемолиза является шизоцитоз, возникающий из-за фрагментации эритроцитов нитями фибрина в тромбированных мелких сосудах (капиллярах, прекапиллярах и концевых артериолах). В крови здорового человека шизоцитов нет, но процедура забора крови и приготовления мазка может приводить к фрагментации самых хрупких клеток, что отражается в ложноположительном шизоцитозе. Поэтому нормальное количество шизоцитов при условии выполнения всех правил подсчета не должно превышать 0,2% общего количества эритроцитов [5, 6].
Из фрагментированных в сосудистом русле эритроцитов высвобождаются лактатдегидрогеназа (ЛДГ) и гемоглобин. Свободный гемоглобин плазмы быстро связывается с гаптоглобином, поэтому его уровень даже при выраженной МАГА в крови остается нормальным, а уровень свободного гаптоглобина снижается. Если же продукция гаптоглобина печенью уменьшается, то в крови может определяться свободный гемоглобин. Этот феномен нередко встречается при HELLP-синдроме и ложно экстраполируется специалистами акушерского профиля на все формы ТМА, даже без поражения печени [7].
При любой ТМА в течение первых двух дней шизоцитов может не быть – именно столько эритроцит может висеть на нитях фибрина без деформации и «почкования». Если сохраняется ТМА-настороженность, подсчет шизоцитов стоит проводить ежедневно [8, 9].
До недавнего времени при возникновении ТМА во время беременности обсуждались только 2 диагноза: тяжелая ПЭ и HELLP-синдром, в том числе его парциальные формы. В то же время в последнее десятилетие было достоверно показано, что такие физиологические гестационные изменения, как снижение активности металлопротеиназы, разрезающей первичные мультимеры фактора Виллебранда на функционально полноценные молекулы малого размера, получившей название ADAMTS13 (A Disintegrin And Metalloprotease with ThromboSpondin 13) и повышение уровня фактора Виллебранда, а также активация системы комплемента создают предпосылки для реализации иных форм ТМА во время беременности и в ранний послеродовый период [10]. Таким образом, во время беременности, помимо HELLP-синдрома и тяжелой ПЭ, возможно развитие любой формы ТМА: прежде всего тромботической тромбоцитопенической пурпуры (ТТП) и комплемент-опосредованной ТМА, которую еще называют атипичным гемолитико-уремического синдромом (аГУС). ТМА также характерна для катастрофического антифосфолипидного синдрома (КАФС). Все эти состояния являются потенциально угрожающими жизни и требуют быстрого решения о выборе тактики лечения.
В связи с тем, что эти заболевания встречаются относительно редко, их проявления практически идентичны, все они могут имитировать тяжелую ПЭ, а терапевтические подходы к ведению таких пациенток различаются, проведение дифференциальной диагностики представляется крайне важным и зачастую определяющим прогноз мероприятием.

Сопоставление частоты различных ТМА в акушерстве
Частота ПЭ варьирует от 3:100 до 7:100 беременностей, достигая в некоторых странах 1:10, что дает основания считать ее самой частой причиной гломерулярного поражения почек [11]. Частота HELLP-синдрома скромнее: по разным данным HELLP и тяжелая ПЭ осложняют 3:1000-1:100 беременностей. Истинная частота ассоциированных с беременностью аГУС (Б-аГУС) и ТТП по мнению исследовательской группы под руководством F.Fakhouri явно недооценена и составляет 1:25 000 беременностей [10, 12]. В общей структуре АФС на долю КАФС приходится не более 1%, однако учитывая распространенность АФС в общей популяции и среди беременных пациенток, можно предположить частоту КАФС 1:100 000 беременностей.
Этиология, патогенез и основные клинические особенности различных форм ТМА, ассоциированных с беременностью
Несмотря на то, что все эти заболевания имеют схожие признаки, характерные для синдрома ТМА в целом, они являются отдельными заболеваниями с разной этиологией и патогенезом.
Тромботическая тромбоцитопеническая пурпура
ТТП – это крайне тяжелое заболевание из группы ТМА, возникающее вследствие недостаточной активности металлопротеиназы ADAMTS13, расщепляющей мультимеры фактора Виллебранда.
Недавнее развитие методов, которые позволяют с высокой точностью измерять активность ADAMTS13, дало возможность диагностировать ТТП в относительно короткие сроки. Считается, что только уровень активности ADAMTS13 ниже 10% высоко специфичен для ТТП, тогда как клинически определяется чрезвычайное разнообразие симптомов болезни. Специфичность снижения уровня ADAMTS13 неоднократно подвергалась сомнениям [13], однако последнее изучение образцов крови и сопоставление клинико-лабораторных изменений у 810 пациентов с ТТП в реестре Великобритании выявило вариации активности металлопротеиназы от 0 до 11%, что подтверждает высокую специфичность этого параметра для данного заболевания [14, 15]. Поэтому до начала терапии плазмой всем больным с ТМА необходимо произвести забор крови для исследования активности ADAMTS13.
Считается, что ТТП может возникнуть на любом сроке гестации, т.к. с момента имплантации уровень ADAMTS снижается практически в два раза, а количество фактора Виллебранда увеличивается почти в 5 раз. Наиболее часто ТТП диагностируется с 20 по 34 неделю [10, 12]. Наибольшим числом наблюдений развития ТТП во время беременности обладает университет Миссисипи: они собрали все литературные описания ТТП во время беременности с 1955 по 2006 гг. – 166 случаев [16]. ПЭ была выявлена у 29 из них (17%), и именно эти женщины имели крайне неблагоприятный прогноз даже при сравнении с изолированной ТТП – материнская смертность, несмотря на плазмотерапию, составила 44,4% против 21,08% (p<0,02). Дифференциация между ПЭ/HELLP-синдромом и ТТП, ассоциированной с беременностью, крайне сложна, но важна для того, чтобы предпринять своевременную и, возможно, спасительную плазмозаменную терапию для излечения ТТП еще до получения результатов исследования ADAMTS13. Поэтому попытки выявить универсальный маркер привели S. Keiser и соавт. (2012) к предположению, что таким маркером может быть отношение ЛДГ к АСТ. В его исследовании у пациенток с HELLP-синдромом (n=83) соотношение ЛДГ к АСТ было значительно ниже 20, тогда как при ТТП отношение ЛДГ к АСТ оказалось более 22 [17]. Интересным оказался факт наличия до беременности изолированной тромбоцитопении у 79% больных, которая ошибочно расценивалась как идиопатическая [18].
Развитие ТТП во время беременности крайне опасно для плода. Почти в половине случаев происходит антенататальная гибель. Поэтому при уже установленном до беременности диагнозе ТТП, рекомендуется применение профилактических инфузий плазмы с ранних сроков беременности [18]. Целью плазмопрофилактики является восполнение дефицита ADAMTS 13 до клинической манифестации болезни.
Атипичный ГУС, ассоциированный с беременностью (Б-аГУС).
Атипичный ГУС занимает около 10% в общей структуре ГУС и представляет собой редкое угрожающее жизни заболевание, развивающееся вследствие наследственно-обусловленной или приобретенной потери функции белков-регуляторов комплемента: фактора Н, мембранного кофакторного протеина (МСР), фактора I, фактора В и др. Дефектное функционирование этих белков приводит к неконтролируемой активации комплемента и развитию комплемент-ассоциированной ТМА [19–22].
Заболевание характеризуется крайне неблагоприятным прогнозом: до 75% пациентов даже при первом эпизоде болезни умирают или быстро достигают терминальной почечной недостаточности. Для амплификации комплемента (перехода «подпороговой» холостой активации в «надпороговую») и развития спорадического аГУС необходимо воздействие «пусковых» факторов. По мнению M. Riedl, F. Fakhouri, (2013), одного пускового фактора часто недостаточно для манифестации ТМА, необходимо сочетание нескольких различных триггеров [23]. Значимость различных триггеров, а также вклад каждого в развитие ТМА неясны. По-видимому, люди, имеющие мутации белков-регуляторов, требуют меньшее количество триггеров или пусковые факторы меньшей значимости. Некоторыми учеными выдвинуто предположение, что такие пациенты или не доживают до беременности, или к детородному возрасту имеют неоднократные повторные эпизоды аГУС и необратимо утраченную функцию почек [24]. Развитие аГУС возможно и в отсутствие генетических мутаций белков-регуляторов, но при наличии большего числа пусковых факторов, соответствующих теории «двойного удара» или эквивалентных тромботическому шторму в развитии КАФС, когда каждый последующий триггер усиливает действие предыдущего [23]. Одним из пусковых факторов может быть диарея, наличие которой характерно не только для типичного ГУС. В последнее время широко обсуждается вероятная высокая значимость STEC (шига-токсин-продуцирующей E. coli) как мощного триггера активации комплемента [24].

В 7–30% случаев развитие аГУС связано с беременностью (Б-аГУС). Установлено, что Б-аГУС чаще всего (74%) развивается в III триместре и раннем послеродовом периоде, тогда как на I триместр приходится всего 11% случаев, а на II – до 15% [25]. Б-аГУС характеризуется агрессивным течением и крайне неблагоприятным прогнозом: материнская смертность достигает 42% [20, 26]. Манифестация этого грозного заболевания в послеродовом периоде не кажется случайной: во время беременности на поверхности трофобласта в достаточном количестве экспрессированы белки-ингибиторы комплемента, такие как DAF (decay-accelerated factor) и MCP, что предотвращает локальную активацию комплемента в плаценте даже при наличии системной активации [27]. Напротив, после родоразрешения, мощный выброс тромбопластина с обширной раневой поверхности, отсутствие «комплемент-ингибирующей» плаценты, попадание в материнский кровоток клеток плода, кровотечение, инфекция могут привести к системной активации альтернативного пути. «Комплемент-ингибирующая» функция плаценты крайне важна для нормального развития беременности. Именно предотвращением локальной активации комплемента в плаценте можно объяснить отсутствие синдрома задержки роста и развития плода в ряде случаев при ПЭ. При нарушении экспрессии антикомплементарных белков в плаценте развивается синдром потери плода, характерный, например, для АФС. Привычное невынашивание беременности при «акушерском» АФС по мнению ряда экспертов связано именно с дефектом локальной антикомплементарной защиты плаценты, запускающей комплемент-опосредованное «отторжение фетального аллотрансплантата» [28].
Акушерский катастрофический антифосфолипидный синдром (КАФС)
КАФС – редкий, но практически в 50% случаев фатальный вариант АФС, в основе которого лежит преимущественное поражение сосудов малого калибра, развивающееся в короткие сроки (от нескольких часов до нескольких дней), приводящее к развитию полиорганной недостаточности и ассоциированное с высоким титром антифосфолипидных антител [29]. КАФС может развиться как при первичном (ПАФС), так и при вторичном антифосфолипидном синдроме (ВАФС), то есть в большинстве случаев к моменту манифестации диагноз АФС уже установлен или предположителен. Предположить наличие АФС позволяют анамнестические указания на тромбоз, неоднократные ранние или однократные поздние репродуктивные потери, тяжелую ПЭ/эклампсию или тяжелую плацентарную недостаточность. Кроме того, у таких пациентов может быть указание на персистенцию антифофолипидных антител (АФА) без клинических проявлений. Однако у трети больных КАФС становится первым клиническим проявлением АФС [30]. В клинической картине «классического» или «сосудистого» АФС преобладают артериальные или венозные тромбозы крупных сосудов, «акушерский» АФС характеризуется синдромом потери плода и представляет собой поражение микроциркуляторного русла, что роднит его с «микроангиопатическим» АФС, основным представителем которого является КАФС [31].
Тромботическая окклюзия сосудов микроциркуляторного русла при КАФС формирует полиморфную клиническую картину, чаще всего представленную симптомами поражения паренхиматозных органов, тогда как тромбозы артерий и/или вен большого калибра, характерные для «классического» АФС, развиваются не более чем у 20% больных. Для развития КАФС также характерно наличие триггеров, основным из которых считают инфекционный процесс, поэтому одним из механизмов запуска КАФС считают молекулярную мимикрию [31].
Согласно концепции тромботического шторма, предложенной С. Kitchens в 1998, КАФС и другие заболевания с массивным тромбообразованием развиваются вследствие избыточного ответа на первоначальный протромботический стимул. В основе этого явления лежит прогрессирующая активация образования тромбина и угнетение фибринолиза с потреблением антикоагулянтных факторов, в результате чего развивается выраженная органная ишемия, усугубляющая процесс выбросом провоспалительных цитокинов, которые в свою очередь лишь потенцируют процесс тромбообразования [32].
Морфологической основой КАФС является распространенная ТМА, аналогичная таковой при ГУС и ТТП, а также синдроме диссеминированного внутрисосудистого свертывания (ДВС). В связи с этим на 10-м Международном конгрессе по АФА были приняты классификационные критерии КАФС и введены понятия «достоверного» и «вероятного» КАФС. (табл. 1). К сожалению, «достоверный» КАФС, диагностируемый при наличии всех 4 критериев, чаще всего устанавливается посмертно, так как для этого требуется гистологическое подтверждение ТМА. Следует помнить, что во время беременности плацента является полноценным органом, поэтому морфологическое исследование плаценты после родоразрешения позволит как насчитать необходимое количество органов для диагноза, так и подтвердить ТМА.
Преэклампсия, HELLP-синдром
Патогенез ПЭ сложен и не до конца расшифрован. Установлено, что в основе развития ПЭ лежит нарушение процесса плацентации, приводящее к неполноценной инвазии трофобласта на ранних сроках беременности. В результате плацента развивается в условиях ишемии и в избыточном количестве начинает секретировать мощный антиангиогенный фактор – растворимый рецептор к васкулоэндотелиальному фактору роста (VEGF), идентифицированный как растворимая fms-подобная тирозинкиназа-1 (sFlt-1). Этот фактор ингибирует как сосудистый, так и плацентарный факторы роста (PlGF), обеспечивающие нормальное развитие и функцию плаценты, и, циркулируя в кровотоке матери, может вносить свой вклад в развитие системной эндотелиальной дисфункции, лежащей в основе всех клинических проявлений ПЭ [33, 34].
Морфологическая картина поражения почек при ПЭ, несмотря на крайнюю редкость тромбозов капиллярных петель клубочка, представлена другими признаками острой ТМА: гломерулярным эндотелиозом с отеком эндотелиальных клеток и отслойкой их от базальной мембраны, приводящим к окклюзии просвета капилляров [35, 36].
Сочетание эклампсии, гемолиза и тромбоцитопении впервые было описано E. Stahnke (1922 г.) и лишь спустя 60 лет L.Weinstein (1982 г.) ввел название HELLP-синдрома, тем самым выделив необходимые критерии диагностики заболевания [1]. В настоящее время диагностика данного состояния основана на критериях Tennessee и Missisipi. Первые включают только одну степень тяжести — максимальную: тромбоциты <100 000⁹ /л, АСТ >70 ЕД/л, ЛДГ >600 ЕД/л. По критериям Missisipi выделяют три класса тяжести HELLP-синдрома: 1 класс — тромбоциты <50 000⁹ /л, АСТ, АЛТ >70 ЕД/л, ЛДГ >600 ЕД/л, 2 класс — тромбоциты 50 000–100 000⁹ /л, АСТ, АЛТ >70 ЕД/л, ЛДГ >600 ЕД/л, 3 класс — тромбоциты 100 000–150 000⁹/л, АСТ, АЛТ 40–70 ЕД/л, ЛДГ >600 ЕД/л [37]. Считается, что HELLP-синдром в 10–20% случаев развивается в отсутствие ПЭ, всего лишь 3% пациенток имеют АФС. Морфологическая картина изменений печени при HELLP-синдроме имеет выраженное сходство с изменениями почек при ПЭ и также представлена ТМА: при относительно редком капиллярном тромбозе отмечается перипортальный некроз гепатоцитов с депозитами фибрина в просвете синусоидных капилляров, плазматическим пропитыванием стенок артериол, отслойкой и десквамацией эндотелиальных клеток от базальной мембраны [38]. Таким образом, HELLP-синдром представляет собой преимущественно «печеночный» вариант ТМА.
До сих пор считается, что треть случаев HELLP-синдрома разворачивается в послеродовом периоде. Однако есть все основания для пересмотра диагностической концепции в таких ситуациях. Многочисленные описания послеродового HELLP-синдрома свидетельствуют о катастрофической тяжести последнего: именно за счет этих случаев материнская смертность при HELLP-синдроме даже в современном мире достигает 25% [39], тогда как 20 лет назад достигала 75% [40]. Послеродовый дебют HELLP-синдрома часто сопровождается развитием полиорганной недостаточности с острым почечным повреждением и отеком легких. Понятие «гормональной бури», которым объясняют послеродовую манифестацию, пришло из описания случая послеродового Б-аГУС с высоким уровнем печеночных ферментов, но до сих пор прочно ассоциируется именно с HELLP-синдромом, хотя по сути объясняет многофакторный пусковой механизм активации комплемента [41]. К сожалению, исследование «PIERS» (Preeclampsia Integrated Estimate of RiSk) [42], показавшее достоверную прогностическую значимость в отношении материнской смертности наличия тромбоцитопении, повышения АЛТ, АСТ и повышения креатинина более 110 мкмоль/л, не изучало послеродовый дебют предполагаемого HELLP-синдрома.
При HELLP-синдроме в 18–36% случаев также описаны мутации генов регуляторных белков комплемента [43]. При исследовании спектра генетических аномалий у таких пациентов, наиболее часто выявлялась гетерозиготная мутация MCP (СD 46).
В исследовании F. Crovetto (2012) обнаружена взаимосвязь именно между послеродовым HELLP-синдромом и носительством мутаций белков- регуляторов комплемента, при этом при «дородовой» манифестации HELLP- синдрома мутации отсутствовали [44]. Высказывается предположение, о том, что HELLP-синдром и Б-аГУС могут быть разными стадиями неконтролируемой активации комплемента. На основании общих морфологических и этиологических факторов предполагается единый патофизиологический механизм неисправной регуляции комплемента в развитии как HELLP-синдрома, так и Б-аГУС. Сочетание различных «пусковых» факторов приводит к генерализованной ТМА при HELLP-синдроме. Таким образом, HELLP-синдром, как и ПЭ могут быть триггерами для развития системной комплемент-опосредованной ТМА. В тоже время у подавляющего большинства заболевание ограничивается ПЭ/HELLP-синдромом и разрешается в послеродовом периоде.
Другие ТМА и ТМА-подобные состояния в акушерстве
Следует подчеркнуть, что и другие акушерские состояния могут вызвать МАГА и тромбоцитопению. К ним относятся сепсис и различные ургентные акушерские состояния, осложняющиеся развитием ДВС. В таких случаях следует помнить, что ДВС является лишь синдромом, утяжеляющим течение основного заболевания (маточное кровотечение, отслойка нормально расположенной плаценты и пр.). Однако если проявления ДВС выходят на первый план, прогрессирует полиорганная недостаточность, нарастает шизоцитоз, МАГА и усугубляется тромбоцитопения, а основная причина, приведшая к ДВС, уже купирована, то нельзя исключить развитие комплемент-ассоциированной ТМА, для которой и причина, и ДВС стали «пусковыми» факторами [45].
Кроме того, описано развитие вторичной ТМА при большом числе бактериальных, вирусных и грибковых заболеваний. С другой стороны, тяжелое состояние пациентки акушерского профиля с системной ТМА имеет все предпосылки для присоединения вторичной инфекции. Эксперты полагают, что если пациент находится в коме, ему проводится искусственная вентиляция легких, у него регистрируется высокая лихорадка и есть признаки нарастающей полиорганной недостаточности, то ведущим нужно считать инфекционный процесс [13].
Алгоритм дифференциальной диагностики ТМА в акушерской практике
Разграничение акушерских ТМА усложняется возможным наслоением одного процесса на другой: так, ПЭ и HELLP-синдром могут стать триггерами развития аГУС, в то же время у женщин, перенесших ТТП, во время беременности выше риск развития ПЭ и HELLP-синдрома [46]. Имеется описание развития ТТП с отслойкой сетчатки у женщины через месяц после родоразрешения по поводу HELLP-синдрома в 32 недели, восстановившей гематологические показатели в послеродовом периоде [47].
Длительно существующий алгоритм диагностики тяжелой ТМА до получения результатов ADAMTS 13%, основанный на выраженности тромбоцитопении и азотемии, и во время беременности может служить ориентиром, но не определяющим диагноз признаком. Так, при преобладании тромбоцитопении (менее 30 000) над почечной недостаточностью (креатинин менее 200 мкмоль/л), вероятность ТТП несколько выше, тогда как при более высоких показателях тромбоцитов и креатинина следует предполагать аГУС (табл. 2) [48–50].
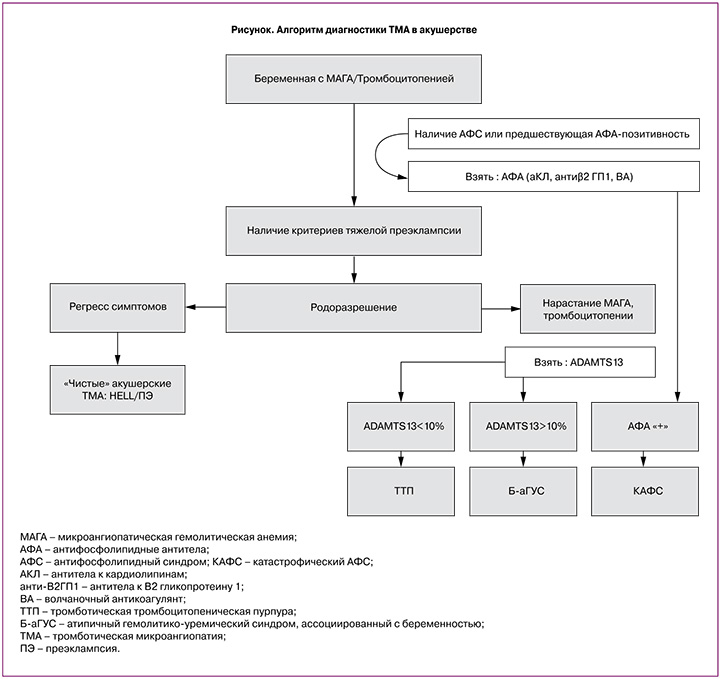
В настоящее время считается, что ключом к дифференциальной диагностике является влияние родоразрешения на регресс ТМА (рисунок): если после родоразрешения МАГА и тромбоцитопения уменьшаются, то можно говорить о «чистых» ПЭ или HELLP-синдроме. Если МАГА и тромбоцитопения сохраняются или нарастают, то следует думать о ТТП и аГУС [51]. При уровне ADAMTS 13 менее 10% устанавливается диагноз ТТП, при ADAMTS 13 более 10% – Б-аГУС. Если имеются признаки фульминантно развившейся полиорганной недостаточности, особенно у пациента с исходным подозрением на АФС, то можно предполагать КАФС. Для его подтверждения необходимы высокие титры АФА.
Терапевтические подходы к акушерским ТМА
По современным представлениям, «чистые» акушерские ТМА, такие как HELLP-синдром и ПЭ, требуют элиминации основного патогенетического фактора-плаценты, которая секретирует мощный антиангиогенный фактор sFlt-1, то есть родоразрешения, поэтому в течение первых 24–48 часов после родов состояние большинства пациентов обычно начинает улучшаться. Терапия кортикостероидами, используемая как до, так и после родоразрешения, не показала своей эффективности для предотвращения материнских и перинатальных осложнений HELLP-синдрома [52]. Ее использование оправдано только для профилактики респираторного дистресс-синдрома новорожденного и при тромбоцитопении менее 50×10⁹/л. Рядом исследователей показана эффективность плазмообмена в лечении беременных с HELLP-синдромом еще до родоразрешения и через 12–24 часа после родоразрешения [53–55]. Очевидно, такая тактика позволяет прервать в целом ряде случаев развитие Б-аГУС и других жизнеугрожающих ТМА, триггером которых может стать HELLP-синдром. Требуются дальнейшие многоцентровые исследования, доказывающие эффективность раннего начала применения экстракорпоральных методов гемокоррекции при HELLP-синдроме, что будет способствовать улучшению клинических исходов.
При нарастании МАГА, тромбоцитопении и прогрессирующей органной дисфункции после родоразрешения следует предполагать Б-аГУС или ТТП, и до получения результатов ADAMTS13 начать инфузии свежезамороженной плазмы или плазмообмен из расчета 30–50 мг/кг веса до стабилизации состояния. Если в течение 2–4 дней регресса не происходит, уровень ADAMTS 13 более 10%, то следует диагностировать Б-аГУС и заменить плазмообмен на терапию экулизумабом – моноклональным антителом к С5 фрагменту комплемента, блокирующим сборку терминального комплекса. Если ADAMTS 13 менее 10%, то диагностируется ТТП и обсуждается присоединение иммуносупрессивной терапии (ритуксимаб, циклофосфан). Если подозревается вероятный КАФС, то к терапии добавляют глюкокортикоиды в высоких дозах, дополнительно также обсуждается возможность усиления иммуносупрессии [56].
Заключение
Во время беременности возможно развитие любой формы ТМА: как «чисто» акушерской (ПЭ, HELLP-синдрома), так и Б-аГУС и ТТП. Предпосылками к этому являются такие физиологические гестационные изменения, как снижение активности ADAMTS13, повышение уровня фактора Виллебранда и активация системы комплемента, в норме уравновешенная экспрессией плацентарных белков-регуляторов. При этом клинические проявления ТМА не являются специфичными для какого-бы то ни было диагноза. ПЭ и HELLP-синдром могут стать триггерами для развития комплемент-опосредованной ТМА (Б-аГУС), а могут остаться «чистыми» или «изолированными» вариантами акушерской ТМА. «Чистые» ПЭ/HELLP разрешаются после родов. Ключевым дифференциальным признаком между «чистыми» акушерскими ТМА и Б-аГУС/ТТП является сохранение симптомов после родоразрешения. Если регресса симптомов после родоразрешения не происходит, необходимо исследовать уровень ADAMTS 13, основные АФА (антитела к кардиолипину, антитела к β2-гликопротеину1, волчаночный антикоагулянт) для исключения/подтверждения КАФС (особенно при поражении 3 и более органов). При геморрагическом колите также рекомендуется исследование STEC (шига-токсин-продуцирующую E.сoli). Уровень сывороточного креатинина более 150–200 мкмоль/л и тромбоцитов более 30 000⁹ практически исключают диагноз тяжелого дефицита ADAMTS13.
При нарастании гематологических проявлений ТМА после родоразрешения необходимо начать плазмотерапию из расчета 30–50 мг/кг с последующей коррекцией ее после получения результатов дообследования.


