
Задержка роста плода (ЗРП), наряду с преэклампсией и преждевременными родами, относится к так называемым «большим акушерским синдромам» [1] – состояниям, ассоциированным с неполноценной плацентацией вследствие нарушений ремоделирования и обструктивных поражений спиральных артерий и вносящим наибольший негативный вклад в структуру перинатальной и материнской заболеваемости и смертности [2].
С учетом значимости указанных состояний с 2021 г. в Российской Федерации внедрен популяционный комбинированный скрининг I триместра беременности [3], направленный на раннее выявление структурных мальформаций, высокого риска хромосомных аномалий и ЗРП, а также случаев с высоким риском преждевременных родов и развития преэклампсии [4].
Неразрывность взаимосвязи системы «мать – плацента – плод» на дородовом этапе обуславливает возможность замедления темпов роста плода как в результате воздействия изолированных либо сочетанных патологических материнских и плацентарных факторов, так и в результате генетических и структурных аномалий самого плода, выявляющихся значимо чаще при ЗРП, в сравнении с когортами нормально растущих плодов [5–8].
Современные критерии антенатальной диагностики ЗРП, утвержденные специалистами в опросе 2016 г. по Дельфийской системе [9], рекомендованные ко всеобщему применению практическим руководством международного общества ультразвука в акушерстве и гинекологии (ISUOG, 2020) [10] и одобренные Министерством здравоохранения Российской Федерации [11], делают отдельный акцент на отсутствие врожденных пороков развития плода для случаев, в которых подобная классификация должна применяться.
Однако даже среди плодов, не имеющих структурных мальформаций, но демонстрирующих замедление роста по данным пренатального ультразвукового исследования, отмечается повышение частоты генетической патологии в сравнении с нормально растущими плодами [7, 8].
Принимая во внимание возможность подобной ассоциации при выявлении плодов с задержкой роста, в особенности ее ранней, диагностируемой до 32 недели беременности, формой [11, 12], рекомендована консультация будущей матери у генетика, обсуждение возможных вариантов и выбор методов генетического обследования [11, 13, 14].
Современные методы генетической диагностики позволяют выявлять не только хромосомные, но и субмикроскопические и моногенные нарушения, которые могут являться причиной ЗРП [6]. С целью выявления генетических аномалий у плода в настоящее время применяется неинвазивный пренатальный скрининг (НИПС) и инвазивная пренатальная диагностика (забор ворсин хориона и амниоцентез) [6, 15]. НИПС является высокочувствительным и специфичным методом скрининга трисомий хромосом 21, 18 и 13, а также анеуплоидий половых хромосом [16, 17], причем последние являются наиболее распространенными у живорожденных детей.
Однако, несмотря на существующие в настоящий момент возможности инструментальных и лабораторных методов обследования беременных, уникальность отдельных клинических случаев позволяет закончить диагностический поиск только в постнатальном периоде.
Клиническое наблюдение
Пациентка 30 лет обратилась в ФГБУ «НМИЦ АГП им. акад. В.И. Кулакова» Минздрава России (далее – Центр) для наблюдения за течением беременности. Из анамнеза известно, что в детстве пациентка перенесла ветряную оспу и краснуху. В 16 лет у пациентки был отмечен единственный судорожный приступ, диагностирована эпилепсия, проводилась терапия вальпроевой кислотой в течение одного года, на момент наступления беременности с амбулаторного учета невролога снята. Гинекологический анамнез отягощен развитием гематокольпоса с момента менархе в связи с полным заращением девственной плевы, потребовавшим хирургической дефлорации.
Данная беременность первая, наступила в результате естественного зачатия в зарегистрированном браке. В I триместре беременность была осложнена рвотой умеренной степени тяжести, проводилось амбулаторное лечение.
При проведении пренатального скрининга в 12 недель беременности ультразвуковое исследование не выявило признаков врожденных пороков развития и хромосомных аномалий у плода. При этом пульсационный индекс в маточных артериях превышал медианные показатели [4] и соответствовал 1,4 значений, кратных медианам (МоМ); среднее артериальное давление у беременной также было повышено, составляя 99,2 мм рт. ст., что соответствует 1,16 МоМ. Напротив, уровни плацентарных гормонов в биохимическом скрининге были ниже медианных показателей, соответствуя 0,34 МоМ для свободной субъединицы бета-хорионического гонадотропина человека, 0,83 МоМ для ассоциированного с беременностью протеина А и 0,58 МоМ для плацентарного фактора роста. Соответственно, по данным комбинированного скрининга был установлен низкий риск трисомий хромосомы 21 (1:11333), 18 и 13 у плода (<1 из 20 000), высокий риск ЗРП (1:97) и преэклампсии (1:24); начата антиагрегантная профилактика реализации данных осложнений низкими дозами аспирина [3].
Для увеличения чувствительности пренатального выявления анеуплоидии у плода в сроке 14 недель пациентка прошла НИПС [18], который выявил высокий риск нарушений по половым хромосомам у плода женского пола. Врачом-генетиком рекомендована инвазивная пренатальная диагностика [19], от которой женщина отказалась, приняв решение о пролонгировании беременности, вне зависимости от возможности наличия аномалий кариотипа плода.
Начиная с 19 недель беременности результаты динамических допплерографических исследований маточно-плацентарного кровотока демонстрировали сохраняющиеся нарушения с выраженным повышением значений пульсационного индекса в маточных артериях, достигающих сотого процентиля и не имеющих тенденции к снижению. Размеры плода не выходили за границы референсных интервалов до 25-й недели беременности, когда была диагностирована ранняя задержка роста плода: предполагаемая масса соответствовала 2 процентилю.
На сроке 24 недели беременности пациентка была госпитализирована в акушерское отделение Центра; в связи с дебютом гестационной артериальной гипертензии на фоне высокого риска реализации преэклампсии, начата антигипертензивная, антикоагулянтная терапия. Несмотря на весь спектр профилактических и лечебных мероприятий, на сроке в 28–29 недель беременности в связи с быстрым нарастанием тяжести преэклампсии (нарастание отеков на фоне снижения темпа диуреза, нестабильные цифры артериального давления с повышением до 170/110 мм рт.ст. при проведении многокомпонентной антигипертензивной терапии) пациентка была переведена в отделение анестезиологии и реанимации для лечения и стабилизации состояния. Однако в связи с дальнейшим нарастанием тяжести преэклампсии, нарастанием отеков с генерализацией и присоединением асцита и выпота в плевральные полости в сроке 29 недель 4 дня было произведено родоразрешение путем операции кесарева сечения в экстренном порядке.
Родилась живая недоношенная девочка с оценкой по шкале Апгар на 1-й минуте – 6 баллов, на 5-й минуте – 7 баллов, массой 900 г (5,3 процентиль, -1.6 Z-score), ростом 32 см (0,4 процентиль, -2.6 Z-score) [20], что соответствует критериям задержки роста для новорожденных, утвержденных Дельфийским консенсусом в 2018 г. [21].
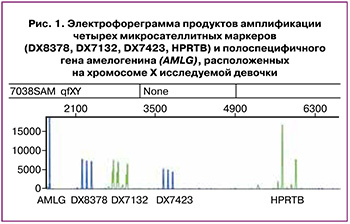
В связи с высоким риском трисомии хромосомы X на основании НИПС, был проведен хромосомный анализ, который показал кариотип 47,XXX. Используя метод количественной флуоресцентной полимеразной цепной реакции, материнское происхождение трисомии хромосомы X определяли путем сравнения коротких тандемных повторов (STR-локусов). У девочки выявлена трисомия по хромосоме X (XXX) по всем маркерам (рис. 1).
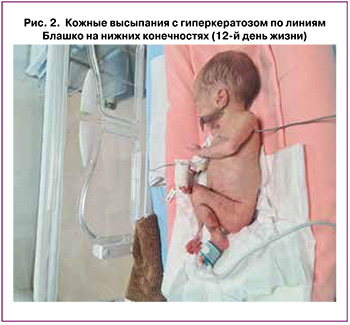
С 1-х суток жизни у ребенка отмечались множественные элементы везикуло-пустулезной сыпи на конечностях, туловище, спине (рис. 2), в связи с чем назначена местная терапия: лосьоны на основе каламина и оксида цинка, декспантенол. На 12-е сутки жизни, кроме везикул, наблюдались бородавчатые высыпания с гиперкератозом по линиям Блашко. Поражения глаз зафиксировано не было.
При повторном тщательном сборе анамнеза было установлено, что аналогичные поражения кожи наблюдались у матери после рождения. Они почти полностью исчезли в возрасте около 6 месяцев. Однако в настоящее время на нижних конечностях сохраняются участки депигментации.
Указанное позволило заподозрить у ребенка недержание пигмента (НП) или синдром Блоха–Сульцбергера [22], для подтверждения которого было проведено молекулярно-генетическое исследование.
Геномную ДНК экстрагировали из лейкоцитов периферической крови с использованием набора для выделения ДНК PREP-MB MAX (Россия).
Анализ методом определения полиморфизма длин амплифицированных фрагментов проводился для распространенной делеции гена IKBKG (NEMO) и включал в себя полимеразную цепную реакцию с последующей детекцией продуктов амплификации на электрофорезе по разнице длин фрагментов норма/мутация.
Учитывая клиническую картину, родословную семьи, данные молекулярно-генетического обследования ребенку был выставлен диагноз: «Недержание пигмента, везикулезная стадия (синдром Блоха–Сульцбергера)». При осмотре неврологом патологической неврологической симптоматики не отмечалось. По данным проведенной нейросонографии головного мозга очаговых изменений выявлено не было. Электроэнцефалография на 8-е сутки жизни показала нормальную картину. Аудиологический скрининг пройден: ребенок не отнесен к группе высокого риска по развитию нарушений слуха. Неонатальный период протекал в соответствии с постконцептуальным возрастом; проводилась комплексная: инфузионная, антибактериальная, противогрибковая, антианемическая, эубиотическая и витаминотерапия, профилактика рахита, бронхолегочной дисплазии и стимуляция дыхательного центра. На 54-е сутки жизни в удовлетворительном состоянии ребенок был выписан домой с массой 2100 г.
Мать девочки консультирована по вопросам правильного ухода и своевременного прохождения с ребенком осмотров смежными специалистами в рекомендованные сроки. Пациентке рекомендованы генетическое обследование, прегравидарное наблюдением у врача акушера-гинеколога и терапевта на этапе планирования будущей беременности.
Обсуждение
Как известно, ЗРП может быть вызвана материнскими, плацентарными, фетальными или генетическими факторами [23].
По первым опубликованным несколько десятилетий назад данным хромосомные аномалии плода составляли до 19% от общего числа родившихся с ЗРП [24].
Однако на современном уровне ранней антенатальной диагностики и элиминации плодов со структурными пороками и/или синдромальной патологией аномальный кариотип выявляется приблизительно у 4% плодов с ЗРП [8].
До недавнего времени клетки плода для проведения кариотипирования получали только с помощью инвазивных манипуляций. Внедрение в практику НИПС в дополнение к биохимическим и сонографическим маркерам может способствовать снижению количества внутриматочных манипуляций, а следовательно, и связанных с ними осложнений [18, 25].
Несмотря на то что скрининг анеуплоидий половых хромосом с помощью НИПС имеет больше ограничений по сравнению с обнаружением трисомий хромосом 21, 18 и 13, он демонстрирует высокую чувствительность и специфичность наряду с умеренной положительной прогностической ценностью [26].
В представленном клиническом наблюдении ранней ЗРП именно НИПС позволил заподозрить аномалию половых хромосом, подтвержденную постнатальным исследованием, выявившим трисомию хромосомы X.
Трисомия хромосомы X – это наличие дополнительной Х-хромосомы у женщин (47,ХХХ вместо 46,ХХ), встречающееся примерно в 1 случае на 1000 рожденных девочек [17]. Поскольку существует вариабельное фенотипическое проявление: от бессимптомного и очень легкого поражения до значительных физических и психологических особенностей, предполагается, что популяционная частота данной патологии установлена крайне неточно: вероятно, только 10% людей с трисомией X знают о своем диагнозе [27, 28].
Трисомия хромосомы X возникает из-за нерасхождения в цикле клеточного деления во время гаметогенеза или после зачатия. Исследования показали преобладание вклада материнских гамет; только 10% случаев были связаны с отцовскими мутациями [29].
Хотя при проведении НИПС кариотип 47, XXX чаще всего является случайной находкой [30], описаны случаи сочетания данного вида анеуплоидии с ЗРП [8].
В более ранних публикациях рядом авторов также было отмечено, что новорожденные девочки с трисомией хромосомы Х имеют меньшую массу тела при рождении и меньшую окружность головы, чем здоровые дети [31].
В описанном нами клиническом наблюдении данный аномальный кариотип ребенка мог внести вклад в развитие ЗРП. Кроме того, нельзя исключить дополнительное негативное влияние сочетания данной патологии с синдромом Блоха–Сульцбергера.
НП, или синдром Блоха–Сульцбергера, является редким Х-сцепленным доминантным генодерматозом с распространенностью примерно 0,7/100 000 живорождений [32, 33].
НП вызывается мутацией гена IKBKG, расположенного в хромосомной области Xq28, кодирующей белок NEMO. Большинство пациентов с НП имеют делецию экзона 4–10 в гене IKBKG [34–39].
Интересным представляется факт, что 65–75% случаев заболевания вызваны спорадическими мутациями, а остальные являются семейными.
Заболевание встречается преимущественно у женщин, поскольку мутация летальна для плодов мужского пола, приводя к антенатальной их гибели еще до начала II триместра беременности. В редких случаях могут выжить плоды мужского пола с сочетанием НП и соматического мозаицизма, либо с синдромом Клайнфельтера. В подобных ситуациях может происходить дальнейшая передача мутации по отцовской линии [32, 33, 40–42].
НП является мультисистемными заболеванием, затрагивающим кожу, глаза, волосы, центральную нервную систему и зубы [22, 32, 33, 43, 44], что требует регулярного мультидисциплинарного наблюдения за больными детьми и взрослыми.
Клиническая картина НП широко варьирует даже у близких родственников при семейном типе наследования [36]. Ключом к диагностике НП, как в описываемом случае, являются специфические кожные проявления, которые могут развиваться как классически – в 4 стадии: (1) эритема и везикопустулы; (2) гиперкератотические, бородавчатые папулы и бляшки; (3) гиперпигментированные пятна; и (4) гипопигментированные атрофические бляшки с алопецией, так и возникать одновременно [37, 38]. К примеру, в представленном клиническом наблюдении на момент выписки у ребенка были перекрывающиеся признаки везикулобуллезной и веррукозной стадий.
В отечественной и зарубежной литературе опубликованы единичные случаи постнатальной диагностики НП, и, хотя в большинстве из них дети имели массу, соответствующую сроку гестации [45–52], в одном из случаев наследования мутации от отца девочка с НП родилась в 36–37 недель беременности с массой 2079 г (-1,3 SD) и ростом 43,5 см (-1,4 SD) [40].
Особое внимание обращает на себя случай пациентки с НП, чья беременность осложнилась антенатально диагностированной, постнатально патоморфологически подтвержденной плацентарной дисфункцией и закончилась родами в 36 недель беременности в связи с преждевременным излитием околоплодных вод. У девочки массой 2640 г, ростом 47 см, родившейся с умеренно выраженной неврологической симптоматикой за счет перенесенной внутриутробной гипоксии, с наличием на коже множества пузырей с прозрачным содержимым, был впоследствии подтвержден диагноз НП [53].
В приводимом нами клиническом наблюдении существует высокая вероятность семейной формы наследования синдрома Блоха–Сульцбергера от матери, имеющей с момента своего рождения характерные кожные проявления, а также анамнез, отягощенный патологией нервной, репродуктивной систем, осложненным течением беременности.
Важно отметить, что для пациенток с НП репродуктивного возраста характерна высокая частота самопроизвольных прерываний беременности, вызванных как гибелью плодов мужского пола, имеющих мутацию, так и частотой различных анеуплоидий, выявляемых у 50–70% эмбрионов при проведении преимплантационного генетического тестирования в программах вспомогательных репродуктивных технологий [54, 55].
На настоящий момент опубликовано только одно сообщение о живом ребенке с сочетанием НП и анеуплоидии – трисомии хромосомы 21 [56].
Сообщений о диагностированном сочетании НП с трисомией хромосомы X у ребенка с задержкой антенатального роста до настоящего времени не было, в связи с чем прогнозирование отдаленных клинических проявлений является затруднительным.
В норме у женщин происходит случайная инактивация одной Х-хромосомы. У большинства женщин с НП наблюдается неслучайная инактивация одной Х-хромосомы в крови и фибробластах из-за потери клеток с мутацией [57].
Наличие сверхчисленной Х-хромосомы может приводить к нарушению дозы генов, избегающих инактивации; поэтому возможно, что у нашего пациента будут более легкие клинические проявления НП; однако этот эффект может быть ограниченным.
У женщин с трисомией хромосомы X чаще наблюдаются позднее менархе и сниженная фертильность [58], однако опубликованы и противоположные случаи [59].
Гормональные обследования таких пациенток часто свидетельствуют о недостаточности яичников, повышенном риске аутоиммунных заболеваний, в том числе поражающих щитовидную железу; поэтому необходимо тщательное наблюдение гинеколога и эндокринолога.
Кроме того, опубликованы данные о связи трисомии хромосомы Х с нарушениями церебральных структур и патологическими отклонениями в высшей нервной деятельности [60–62].
Важно отметить, что ЗРП, недоношенность с экстремально низкой массой тела при рождении, даже в изолированном от генетических патологий виде, являются факторами высокого риска развития ранних и отдаленных кардиоваскулярных, неврологических, офтальмологических и эндокринных осложнений у ребенка, приводящих к снижению качества и сокращению продолжительности предстоящей жизни [63–65].
Для комплексного ухода за нашими пациентками – матерью и ребенком решающее значение будет иметь долгосрочное и тесное междисциплинарное сотрудничество между акушерами-гинекологами, дерматологами, педиатрами, неврологами, эндокринологами и генетиками [66–68].
Заключение
Данное клиническое наблюдение впервые в мире выявленного сочетания трисомии хромосомы Х и НП у ребенка с внутриутробно развившейся ранней задержкой роста демонстрирует возможности и ограничения современных инвазивных и неинвазивных методов анте- и постнатальной диагностики сочетанной генетической патологии.
Сложное, на настоящий момент, возможно, непредсказуемое влияние сочетания генетических и перинатальных факторов на последующее развитие ребенка требует междисциплинарной коллегиальности медицинского и педагогического сопровождения его жизни.
Своевременная диагностика генетической патологии у плода, ребенка, взрослого пациента необходима для оказания пациентам специализированной медицинской помощи, снижения показателей заболеваемости, инвалидизации и смертности населения страны и мира в целом.


