
Детский церебральный паралич (ДЦП) является серьезным неонатальным дефектом и значительным экономическим и социальным бременем, который затрагивает 2-3.5 живорождений на 1 000, с распространенностью 17 000 000 по всему миру [1—6]. По данным одного из китайских исследований, распространенность ДЦП составила 2,37 на 1 000 живорожденных в провинции Хэнань [3]. Клинические характеристики ДЦП неоднородны; несмотря на различную манифестацию, общим для разных случаев является непрогрессирующая дисфункция позы и управления движением. ДЦП может быть разделен на различные неврологические подтипы на основе того, какие именно конечности поражены, а также по степени наблюдаемых двигательных нарушений: спастическая диплегия, спастическая тетраплегия, спастическая гемиплегия, дискинетический или дистонический, атаксический, гипотонический, или смешанный. На основную подгруппу — спастических (диплегических, тетраплегических или гемиплегических) ДЦП — приходится 70—80% всех случаев ДЦП [1, 2].
Эпидемиологические исследования показали, что более одной трети младенцев с ДЦП родились в результате преждевременных родов, а еще 15% имели острую энцефалопатию после своевременных родов [7, 8]. Ранее считалось, что неблагоприятные факторы внешней среды, приводящие к асфиксии в родах, являются общими факторами для развития ДЦП. В настоящее время, однако, считается, что в большинстве случаев ДЦП развивается вследствие внутриутробного повреждения развивающегося мозга, и лишь некоторые случаи обусловлены исключительно тяжелой гипоксией или ишемией в родах. Многими исследователями признано, что ДЦП обычно возникает из-за внутриутробного нарушения (пренатально) в развитии головного мозга, особенно между 24-й неделей гестации и родами. Как генезис, так и неблагоприятные перинатальные клинические условия могут вносить свой вклад в этиологию ДЦП [1, 2]. Некоторые исследования позволяют предположить, что гипоксическая ишемия или эффекты пренатальной инфекции или воспаления могут привести к общему пути перинатальной травмы головного мозга, характеризующейся нейрональной эксайтотоксичностью, апоптозом и микроглиальной активацией в критический период развития. Между тем, все большее число свидетельств указывает на то, что генетические факторы могут быть ответственными за большое количество случаев ДЦП.
В этом мини-обзоре, исследуя последние идеи и выводы о патогенезе ДЦП, мы систематически рассмотрим комплексную этиологию ДЦП, особенно гетерогенность, лежащую в основе генетических вариантов, ассоциированных с этим состоянием или являющихся его причиной.
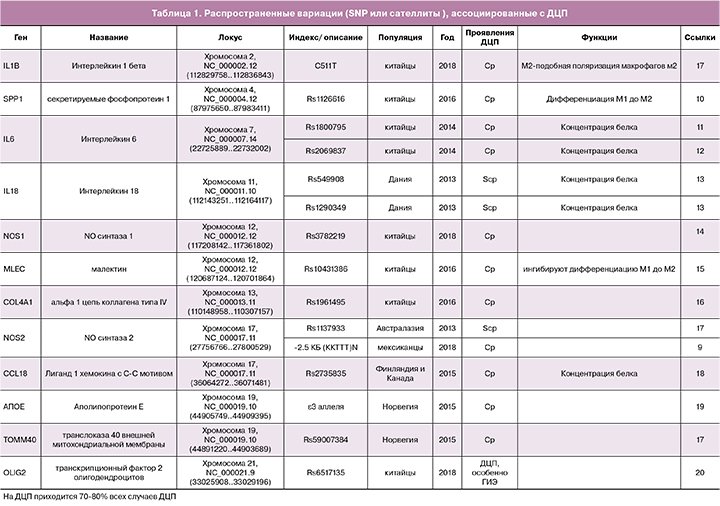
Ассоциация мутаций или изменчивости генов с ДЦП
Генетические исследования семейных и спорадических случаев ДЦП были проведены в основном для изучения изменений в уровнях последовательности ДНК. Большинство исследований генетической восприимчивости к ДЦП наиболее распространенных типов вариабельности, таких, как однонуклеотидные полиморфизмы (SNPs) и микросателлиты, проводились, как исследования ассоциаций с использованием аллель-специфичных микроматриц или масс-спектрометрии, или прямым секвенированием [9—20]. Например, Torres-Merino et al. изучали, повышается ли риск ДЦП при увеличении числа копий 2,5 тысяч пар нуклеотидов (ССТТТ)n - микросателлита в гене NOS2 и при замене C511T в промоторе гена IL-1B у пациентов с ДЦП после перинатальной гипоксическо-ишемической энцефалопатии [9]. Они обнаружили, что образованный в результате этого гаплотип (CCTTT)14/TT, может быть полезным маркером для идентификации пациентов с высоким риском развития ДЦП вследствие гипоксически - ишемической энцефалопатии [9]. В исследовании «случай-контроль» 652 пациентов с ДЦП и 636 здоровых индивидуумов группы контроля в популяции китайцев Хань, рассматривавшем подверженность носителей генетических вариантов NOS1 ДЦП с помощью метода MassARRAY, была выявлена связь полиморфных локусов rs10774909, rs3741475, и rs2682826 с неонатальной энцефалопатией, тогда как rs3782219 был негативно связан со спастической квадриплегией (ОШ = 0,742) [14]. Полиморфные локусы rs10431386 и rs7964786 гена MLEC, как предполагается, участвуют в патогенезе ДЦП у китайской популяции (ОШ составляет 1,587 и 1,956, соответственно). Аллели C этих локусов ингибируют экспрессию MLEC в крови пациентов с ДЦП и в линии макрофагов [15]. В 2015 г. Kallankari H et al. обнаружили, что распространенный вариант локуса rs2015086 в гене CCL18, помимо внутрижелудочкового кровоизлияния, обладает аддитивным влиянием на подверженность ДЦП детей с очень низким гестационным возрастом (менее 32 недель) из северной и центральной Финляндии (25 наблюдений, группа контроля – 195 человек) [18]. Sun L et al. недавно изучали когорту из 763 младенцев популяции Хань с ДЦП и 738 здоровых для поиска ассоциации полиморфизма гена OLIG2 с ДЦП [20]. Они обнаружили незначительную ассоциацию rs6517135 с ДЦП на уровне генотипа, причем эта ассоциация оказалась значительно сильнее в подгруппе младенцев с ДЦП, которые страдали гипоксическо-ишемической энцефалопатией после рождения [20].
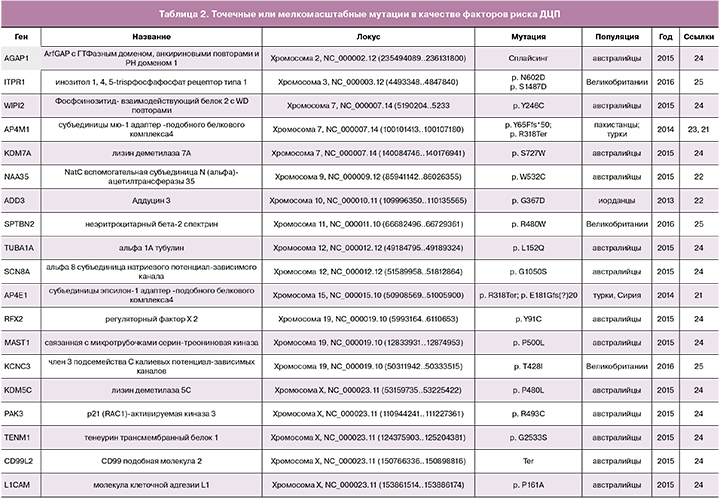
Исследования, основанные на недавно разработанных методах секвенирования, помогли выявить больше de novo мутаций, вероятно, являющихся патогенными. Для изучения ДЦП было использовано высоко производительное полноэкзомное секвенирование, которое является эффективной стратегией для поиска редких вызывающих заболевания мутаций [21—25]. В 2013 г. Kruer M.C. et al., путем картирования гомозиготности и экзомного секвенирования, идентифицировали гомозиготную мутацию c. 1100 G>A (p. G367D) в гене ADD3 [22], кодирующем гамма-аддуцин у всех больных членов иорданской семьи, практиковавшей множественные близкородственные браки. Исследуя in vitro функциональные последствия мутации с помощью полученных от пациентов фибробластов, авторы обнаружили, что она ухудшает актин-кэпирующую функцию аддуцина, что приводит к ненормальной пролиферации и подвижности культивируемых фибробластов. Используя методику полноэкзомного секвенирования у двух братьев с ДЦП, обнаружили новую гомозиготную мутацию гена AP4M1 c. 194_195delAT (p. Y65Ffs*50). Помимо мягких дисморфических черт, умственной неполноценности, спастического парапареза и уменьшенной окружности головы, эти пациенты демонстрируют агрессивное поведение [23]. McMichael G et al. с помощью полноэкзомного секвенирования выявили и подтвердили у 43 из 98 (44%) трио «пациент – родители» 61 de novo вариант, проявляющийся в структуре белков. Десять из них в трех ранее идентифицированных генах-кандидатах (TUBA1A [n=2], SCN8A [n=1] и KDM5C [n=1]) и в шести новых генах-кандидатах (AGAP1, JHDM1D, MAST1, NAA35, RFX2 и WIPI2) были потенциально патогенными для развития ДЦП, тогда как для четырех гемизиготных вариантов на хромосоме X в двух известных генах (L1CAM и PAK3) и в двух новых генах-кандидатах (CD99L2 и TENM1) была предсказана патогенность для развития ДЦП [24]. В 2015 г. Schnekenberg R et al., используя таргетное высокопроизводительное секвенирование или секвенирование экзомов трио «ребенок-родители», выявили у пациентов с атаксическим ДЦП de novo точечные мутации в 3 различных генах — KCNC3, ITPR1 и SPTBN2 [25]. Генетическое тестирование также помогает отличить ДЦП от других заболеваний. Например, для доброкачественной наследственной хореи, вызванной мутациями в гене NK2-гомеобокс-содержащем гене 1 (Nkx2-1), характерны клинические признаки, сходные с таковыми при атаксическом и дискинетическом ДЦП, что приводит к возможности ошибочного диагноза. В 2013 г. McMichael G et al. [26] обнаружили в семье, которой ранее неверно диагностировали атаксический дискинетичесий ДЦП, делецию протяженностью 7 пар нуклеотидов в гене Nkx2-1. Атаксический ДЦП подозревался у отца и двоих его детей до тех пор, пока дети не достигли подросткового возраста, когда появилось предположение о наличии у них доброкачественной семейной хореи. Кроме того, ошибочный диагноз ДЦП может быть поставлен больным с наследственной спастической параплегией на основании общей для этих состояний спастичности нижних конечностей, несмотря на то, что обычно у таких больных есть, как характерные клинические признаки, так и вполне определенные генетические признаки [27, 28].
Различные неврологические заболевания характеризуются изменением количества копий (copy number variation, CNV), которое обычно называют дупликацией или делецией фрагментов ДНК. К ним относятся: синдром дефицита внимания и гиперактивности, аутизм, биполярное расстройство, эпилепсия, тяжелая умственная отсталость и шизофрения. Также для CNV показана связь с ДЦП. Картирование генов-кандидатов в семейных случаях ДЦП проводили с помощью анализа сцепления [29]. Также для рассмотрения роли CNV в развитии ДЦП использовали сравнительный анализ с помощью полногеномных SNP-микроматриц (таблица 3). У 7% из 147 канадских семей с различными подтипами ДЦП (в том числе 37,4% с гемиплегией) были выявлены de novo CNV. Выявленные de novo CNV затрагивают гены GRIK2, LAMA1, DMD, PTPRM и DIP2C, которые, возможно, участвуют в развитии головного мозга. Эти вариации могут вносить вклад в этиологию 18/97 (18,6%) случаев ДЦП [30, 31].

Рассматривая опубликованные исследования, мы подводим итог генетических локусов, которые связаны с ДЦП, в таблицах 1–3. Общая изменчивость на нескольких хромосомах, в том числе 2, 4, 7, 11, 12, 13, 17, 19 и 21, может быть связана с ДЦП; точечные или малые мутации выявлены на 1, 2, 3, 7, 9, 10, 1, 12, 14, 15, 19 и Х хромосомах; релевантные для ДЦП CNV обнаружены почти во всех хромосомах.
Эпигенетическая регуляция как этиология ДЦП
Метилирование ДНК является одним из наиболее важных аспектов эпигенетического регулирования, которое может быть, по крайней мере, частично наследственным. Анализ профиля метилирования ДНК при ДЦП был выполнен с помощью бисульфитного секвенирования уменьшенной представленности в полногеномном масштабе. Эпигеномный анализ пятен крови гомзиготных близнецов, дискордантных по ДЦП, показало заметные различия в метилировании некоторых регионов генома [32, 33]. Кроме того, предложена обладающая, как хорошей специфичностью, так и хорошей чувствительностью диагностическая модель, основанная на полногеномном профиле метилирования, для различия детей со спастическим ДЦП от здоровых детей без ДЦП [34].
Важными участниками эпигенетического регулирования считаются некодирующие РНК. Исследования показали, что микроРНК (miRNAs или miRs), участвующие в такой регуляции, отражают патогенез травмы белого вещества, приводящей к когнитивным нарушениям, поведенческим расстройствам и ДЦП [35]. В 2018 г Li H. et al. [36] обнаружили взаимосвязь между функциональным полиморфизмом в кодирующей области miRNA и предрасположенностью китайских детей к ДЦП. Уровень зрелой miR-124 понижается in vitro при замене С на Т в одном из локусов, а снижение уровня miR-124 приводит к менее эффективному ингибированию генов-мишеней ITGB1, LAMC1 и BECN1, которые могут играть важную роль в развитии нервной системы [34].
Исследования экспрессии генов при ДЦП
Генетические и эпигенетические изменения могут непосредственно изменять экспрессию соответствующих генов. Кроме того, при изучении экспрессии генов необходимо учитывать сложные механизмы геномного регулирования. Удаленный энхансер гена DMRT3 расположен примерно в 120 тысяч пар нуклеотидов дистальнее цис-регуляторного элемента сайта связывания RAR/RXR-в хромосомном регионе 9p 24.3. Ранее было показано, что делеция этого региона приводит к развитию врожденных нейродегенеративных заболеваний, напоминающих ДЦП. При этом, вначале развивается врожденная гипотония, далее переходящая в спастическую квадриплегию, что сопровождается тразиторными нистагмами на первом году [29]. Было также установлено, что у лошадей Dmrt3 может работать, как координатор локомоции, а у мышей уровень его экспрессии высок в развивающемся переднем мозге и в некоторых вставочных нейронах спинного мозга. Эти факты позволяют предположить, что делеция цис-регуляторного элемента DMRT3 у человека может вызвать нарушение развития переднего мозга, приводя к спастическому ДЦП [37]. Ткани детей, больных ДЦП, были изучены на предмет транскрипции генов в масштабе полного генома [38, 39]. Предыдущее исследование показало, что транскрипционные профили спастических мышц пациентов с ДЦП не характерны для других заболеваний, таких, как мышечная дистрофия Дюшенна или мышечная атрофия, вызванная неподвижностью [38]. Еще одно исследование показало, что большинство транскриптов с измененным уровнем экспрессии связаны с более выраженным при ДЦП внеклеточным матриксом и снижением уровня метаболизма и активности убиквитинлигазы, причем усиление внеклеточного матрикса коррелирует с инвалидностью [39]. Также, в некоторых работах измеряли уровни белков, главным образом, для функциональных исследований, основанных на генной изменчивости [11–13, 18] или для скрининга диагностических маркеров. Хотя дети с ДЦП демонстрировали увеличенные энергозатраты при движении, специфичная для типов волокон активность сукцинат-дегидрогеназы, отражающая окислительную способность митохондрий скелетных мышц мощностей, сходна у детей с ДЦП и здоровых детей без ДЦП [40].
Пренатальные воздействия как факторы риска развития ДЦП
В исследовании оценки частоты возникновения ДЦП в группе риска спонтанных преждевременных родов, стратифицированной на основании гестационного возраста, было обнаружено, что риск развития ДЦП снижается с ростом гестационного возраста [40]. Помимо генетического аспекта, на развитие ДЦП могут влиять плацентарная патология, внутриутробная инфекция, материнские заболевания во время беременности (инфекционные, болезни сердца, гипертония, анемия, диабет и заболевания почек), осложнения беременности, и даже загрязнение окружающей среды (воздуха, воды и пищи) [3, 41, 42]. Поэтому интенсивного исследования заслуживает внутриутробное развитие фетального мозга с особым вниманием к синаптической функции.
Заключение
Недавние исследования факторов риска развития и причин ДЦП, особенно новые свидетельства генетических аспектов, улучшили наши знания об этиологии ДЦП. Тем не менее, впереди еще долгий путь к адекватному пониманию ДЦП для снижения заболеваемости.


