
Клинически венозный тромбоэмболизм и рак имеют два основных проявления: с одной стороны тромбоз может быть единственным клиническим симптомом скрыто протекающего рака, с другой – у пациентов с выявленным раком на всех стадиях заболевания может развиться тромбоз. В клинической онкологии тромботические осложнения относятся к наиболее часто встречающимся паранеопластическим синдромам и проявляются артериальными и венозными тромботическими окклюзиями, мигрирующим тромбофлебитом, эмболией легочной артерии, небактериальным тромбоэндокардитом, парадоксальными кровотечениями, тромботической микроангиопатией [1, 2].
Можно выделить три категории факторов риска развития тромбофилии у больных злокачественными новообразованиями. Первую категорию такого рода факторов мы обозначаем как «специфические опухоль-зависимые», вторую – как «общепатологические факторы риска», третью как – «терапия-зависимые» факторы риска.
Патогенез тромбофилии у онкологических пациентов включает факторы, связанные с ответом на опухоль (воспаление, острофазовая реакция, диспротеинемия, очаговые некрозы, гемодинамические нарушения), а также специфические факторы, обусловленные самими опухолевыми клетками и связанными c опухолью макрофагами. А именно: прокоагулянтная и фибринолитическая активность раковых клеток, их взаимодействие с тромбоцитами, мононуклеарными макрофагами и эндотелием, неоангиогенез, лечебные мероприятия (химиотерапия, гормонотерапия). Опухолевые клетки активируют коагуляционную систему или систему фибринолиза, создавая условия для дальнейшего своего распространения, стимуляции ангиогенеза, повышения сосудистой проницаемости, что в свою очередь способствует метастазированию [3–5].
Патогенетические особенности опухолевого роста
В основе патогенеза гемостазиологической паранеоплазии лежит активация как коагуляционного, так и сосудисто-тромбоцитарного звеньев свертывания крови, что обеспечивается [6, 7]:
- нарушением структурной целостности и функциональной стабильности сосудистого эндотелия опухолевыми клетками и цитокинами;
- активацией тромбоцитов опухолевыми клетками, приводящей к их повышенной адгезии и агрегации;
- синтезом прокоагулянтов и ингибиторов фибринолиза опухолевыми клетками;
- прокоагулянтной активностью опухоль-ассоциированных макрофагов и активированных моноцитов периферической крови.
Увеличение прокоагулянтной активности, присутствие всех компонентов системы коагуляции локально в области расположения опухоли и уменьшенная деятельность противосвертывающей системы ведет к гиперкоагуляции как результату злокачественного развития (рис. 1) [8, 9].
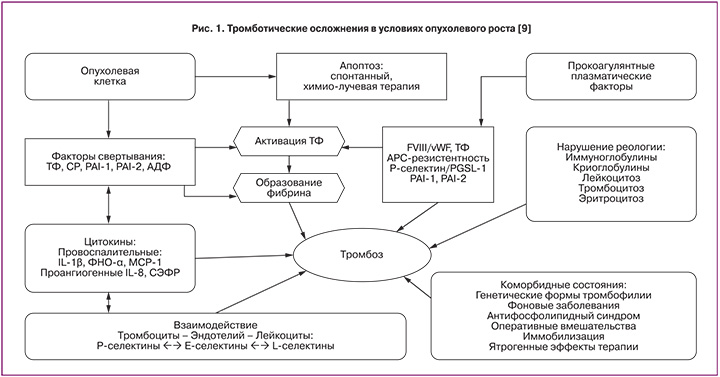
Генерация тромбина и формирование фибрина постоянно обнаруживаются у онкологических пациентов, эти процессы приводят к увеличению риска тромбоэмболических осложнений.
Процесс формирования фибрина является ключевым для роста опухоли и метастазирования. Активация свертывания крови при раке – сложный феномен, включающий множество компонентов системы коагуляции и многочисленные взаимодействия между опухолевыми клетками и клетками крови, включая тромбоциты, моноциты, эндотелиальные клетки [10, 11].
Гемостаз-независимые механизмы опосредуются путем расщепления рецепторов активированных протеаз (PARs) и последующей активацией каскада трансдукции сигнала, связанной с протеином G, которые стимулируют связанные с ангиогенезом гены. Гемостаз-зависимые механизмы опосредуются через осаждение фибрина и активацию тромбоцитов [12].
Действие сосудистого эндотелиального фактора роста (VEGF), и фактора некроза опухоли (TNF-a) также регулирует прокоагулянтную активность тканевого фактора, что ведет к развитию системной гиперкоагуляции, свойственной многим раковым пациентам (таблица) [13, 14].
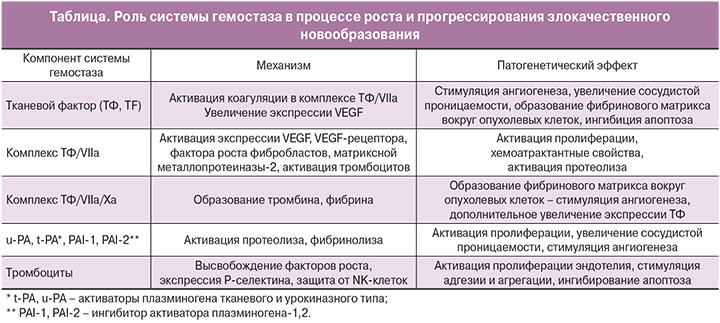
Принципиально важнен тот факт, что тромбофилическое состояние является не просто сопутствующим злокачественному новообразованию процессом, а имеет глубокий общепатологический смысл. Активация системы гемостаза создает условия для роста опухоли за счет опухолевого ангиогенеза и распространения за счет метастазирования раковых клеток [15, 16]. Таким образом, контроль за состоянием системы гемостаза и меры, направленные на профилактику тромбофилии, являются не профилактикой тромбогеморрагических осложнений у онкологических больных, а лечением основного заболевания за счет блокирования путей роста (ангиогенеза) и метастазирования опухоли [17, 18].
При синдроме системного воспалительного ответа наблюдаются признаки активации как внешнего, так и контактного пути коагуляции. Это проявляется в снижении уровня фактора XII и прекалликреина в плазме. Так, эндотоксин и провоспалительные цитокины активируют фактор XII как непосредственно, так и путем обнажения коллагеновых структур, а также активируют калликреиновую систему. Однако установлено, что активация свертывающей системы крови во время сепсиса управляется в основном через внешний путь, опосредованный тканевым фактором. Антитела к тканевому фактору или фактору VII/VIIa либо лечение с помощью TFPI (ингибитор тканевого пути свертывания) предупреждает активацию обоих путей свертывания у обезьян с эндотоксинемией и сепсисом, в то время как антитела к фактору XII не вызывают такого эффекта [19, 20].
Источники тканевого фактора могут различаться в зависимости от патогенеза воспалительного процесса; при новообразованиях одним из основных источников тканевого фактора является сама опухолевая ткань, равно как и СР (раковый прокоагулянт). Установлено, что многие виды опухолей продуцируют и выделяют в кровь большое количество тканевых факторов, а также особых «раковых прокоагулянтов», обладающих способностью активировать как фактор VII, так и фактор X [21].
На рис. 2 представлена принципиальная схема патогенеза тромбофилии у онкологических больных в модификации триады Вирхова.
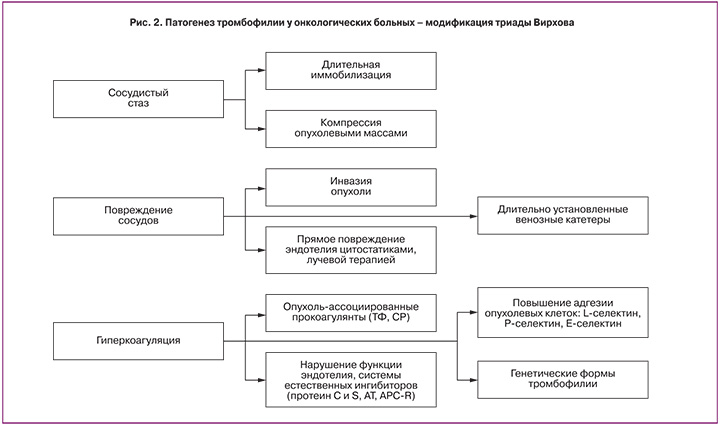
Злокачественная опухоль, являясь, с одной стороны, источником гиперпродукции тромбопластических веществ, «запускает» синдром диссеминированного внутрисосудистого свертывания (ДВС), а с другой, способствует оседанию окутанных фибрином опухолевых клеток в микроциркуляторном русле и возникновению метастазов [22, 23].
Роль нарушений функции гемостаза в процессе опухолевого неоангиогенеза
Ангиогенез обусловливает образование комплексов кровеносных сосудов, которые являются аберрантными и имеют извращенную реологию. Фактически, поток в таких сосудах может изменяться не только по величине, но также и по направлению.
Вслед за повреждением тканей происходит экстравазация фибриногена из кровеносных сосудов в экстраваскулярное пространство с образованием фибринового матрикса. Воспалительные клетки и эндотелиальные клетки мигрируют в такой матрикс и стимулируют процессы репарации. Во-вторых, через какое-то время гель фибрина преобразовывается в зрелую васкуляризированную соединительную ткань путем активации ангиогенеза. В-третьих, фибрин имеет существенное влияние на воспалительную инфильтрацию опухоли, заключающееся в регулировании формирования стромы и защиты опухоли от иммунной системы [24–26].
Образование стромы вокруг опухоли является ключевым моментом в развитии кровоснабжающей ее сосудистой сети. Экстравазация фибриногена из поврежденных сосудов также возможна в процессе развития опухоли. Экстраваскулярный плазменный фибриноген и фибронектин плазмы через коагуляционные эффекты инициируют образование опухолевыми клетками тканевого фактора и в результате – фибриновой сети. Этот фибриновый матрикс стимулирует миграцию воспалительных клеток, тромбоцитов и эндотелиальных клеток, что поддерживает васкуляризацию [26, 27]. Факторы ангиогенеза (например, сосудистый фактор роста эндотелиальных клеток – СФРЭ) экспрессируются в поврежденных тканях и опухолевых клетках. Развитие сосудистой сети, питающей опухоль, требует взаимодействия между эндотелиальными клетками, воспалительными клетками, фибробластами, протеазами, факторами ангиогенеза и временным (провизорным) фибриновым матриксом. Таким образом, «окружение» раневой ткани и развивающейся опухоли имеют схожую природу и поддерживают процесс ангиогенеза [28, 29].
Под действием опухоли эндотелий претерпевает следующие изменения: активация эндотелиальных клеток (1), деградация базальных мембран эндотелия, экссудация фибрина (2), миграция эндотелиальных клеток и пролиферация на новом месте (3), формирование капилярной сети в опухоли (4), образование сосудов (5). В качестве активаторов эндотелия выступают факторы роста: VEGF; bFGF – фактор роста фибробластов; PDGF – фактор роста тромбоцитов; PIGF – плацентарный фактор роста и медиаторы воспаления: IFN – интерферрон, IL – интерлейкины, TNF-α. Система плазмина и его активаторов (plasmin, uPA, uPAR) и система мембранных металлопротеиназ участвуют в деградации базальных мембран, обеспечивая инвазию опухоли и ангиогенез [30, 31].
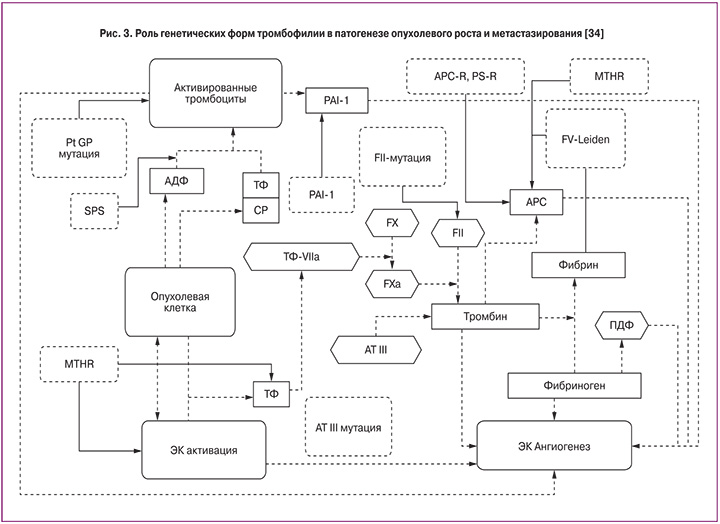
Уже не вызывает сомнений, что антифосфолипидный синдром (АФС) и циркуляция антифосфолипидных антител (АФА) играют ведущую роль в структуре тромбозов, обусловленных патологией гемостаза. Будучи самым распространенным тромбофилическим дефектом гемостаза, АФС может значительно усугублять уже имеющуюся тромбофилию. В последнее время одна из ведущих ролей в патогенезе АФС отводится аннексину V, обладающему мощными антикоагулянтными способностями. АФА, связываясь с фосфолипидами, или протеин-фосфолипидными комплексами, которые могут содержать β2-гликопротеин-1, протромбин или другие протеиновые кофакторы, влияют на способность аннексина V закрывать фосфолипидную поверхность, и, следовательно, усиливают способность фосфолипидов к коагуляционным реакциям [32, 32]. На рис. 3 показано влияние генетических форм тромбофилии на патогенез опухолевого неоангиогенеза и метастазирования [34].
Заключение
Из вышеуказанных механизмов взаимодействия тромбоцит-эндотелий-опухолевая клетка следует, что при наличии в организме опухолевой ткани, с одной стороны, запускается ряд механизмов, направленных на борьбу с дальнейшим распространением неоплазии, а с другой – происходят процессы ангиогенеза и фибринизации опухолевой ткани, что способствует ее росту и метастазированию.
Таким образом, можно считать патофизиологически обоснованным утверждение, что ДВС-синдром в той или иной степени в 100% сопутствует опухолевому росту. Однако же клинически далеко не всегда ДВС-синдром проявляется, из чего многие делают вывод о необязательности профилактики ДВС-синдрома у раковых пациентов, что неизбежно ведет к тромботическим, и реже – к геморрагическим осложнениям.
Вышеописанные патофизиологические особенности делают рак, возможно, наилучшим примером приобретенной тромбофилии. Улучшение понимания патофизиологии тромбофилии при раке должно обеспечить клиницистов точным обоснованием более агрессивных и определенных стратегий противотромботического лечения больных злокачественными опухолями.


