
Актуальность. Тромботическая микроангиопатия (ТМА) – клинико-морфологический синдром, в основе которого лежит повреждение эндотелия сосудов микроциркуляторного русла, вызванное разными причинами, но проявляющееся сходной клинической симптоматикой и гистологическими признаками. Морфологическая картина ТМА – отек эндотелиальных клеток, их отслойка от базальной мембраны (эндотелиоз), некроз, деструкция, расширение субэндотелиального пространства, тромбы в просвете капилляров и артериол, содержащие тромбоциты и фибрин, приводящие к полной окклюзии сосуда. В настоящее время ТМА рассматривается как неотъемлемая часть патогенеза тяжелых акушерских осложнений – преэклампсии и HELLP-синдрома.
Классическими вариантами ТМА являются типичный гемолитико-уремический синдром и его разновидность – аГУС (атипичный гемолитико-уремический синдром), тромботическая тромбоцитопеническая пурпура (ТТП), или болезнь Мошковица. Во время беременности возможно развитие вторичной ТМА – при тяжелой преэклампсии и HELLP-синдроме. Клинические и лабораторные критерии для постановки диагноза ТМА включают:
- наличие микроангиопатической гемолитической анемии (МАГА) – Кумбс-негативная гемолитическая анемия с высоким уровнем лактатдегидрогеназы (ЛДГ), низким уровнем гаптоглобина и наличием шизоцитов в мазке периферической крови);
- тромбоцитопению (потребления);
- ишемическое поражение органов, в первую очередь ЦНС, почек.
При появлении клинических симптомов острой ТМА во время беременности для определения тактики ведения и прогноза применяется дифференциальная диагностика между тяжелой преэклампсией (ПЭ) и HELLP-синдромом, ТТП, ГУС (типичным и атипичным) и КАФС.
ТТП – системная форма ТМА, в основе которой лежит тромбообразование в микроциркуляторном русле, обусловленное сверхкрупными мультимерами фактора фон Виллебранда. Распространенность составляет 0,37–0,45 на 100 000 населения. 70% больных – женщины. Характерным признаком ТТП является дефицит плазменной протеазы, расщепляющей мультимеры фактора Виллебранда – это ADAMTS-13. При семейных формах ТТП наблюдается наследственный дефект этого фермента. В норме металлопротеаза ADAMTS-13 расщепляет крупные мономеры фактора Виллебранда, обусловливая их свободную циркуляцию в сосудах микроциркуляторного русла. При дефиците ADAMTS-13 на нерасщепленных молекулах фактора Виллебранда тромбоциты фиксируются и разрушаются, что приводит к микрососудистому гемолизу – ТМА. Для ТТП характерен резкий дефицит ADAMTS-13 – менее 5%. ГУС типичный – инфекционно обусловленный действием токсина (веротоксин) кишечной палочки (E. сoli О 157:H7) или шига-токсина Shigella dysenteriae. Шига-токсин приводит к прямой активации комплемента с повреждением эндотелия и развитием ТМА. Это эндемичное заболевание, поражающее преимущественно детей. Характерным признаком является развитие почечной недостаточности (олиго- или анурия, гематурия), рис. 1.
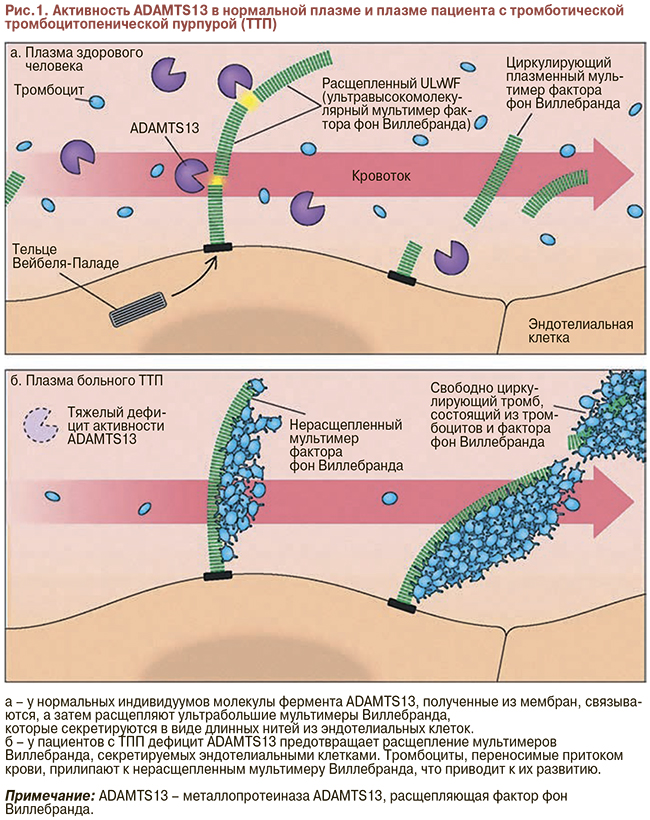
АГУС – это вариант комплемент-опосредованной ТМА, в основе которого лежит хроническая неконтролируемая активация комплемента, ведущая к генерализованному тромбообразованию в сосудах микроциркуляторного русла. Причина – генетически обусловленный дефект в системе комплемента, приводящий к его активации и повреждению эндотелия.
При беременности ТМА может быть обусловлена тяжелой ПЭ и HELLP-синдромом. Более того, впервые аГУС и ТТП могут манифестировать во время беременности. Этими факторами затрудняется дифференциальная диагностика ТМА при беременности.
Дифференциальную диагностику ТМА при беременности следует проводить, опираясь на следующие клинические и лабораторные признаки: уровень тромбоцитопении, артериального давления, наличие коагулопатии, абдоминальные, симптомы поражения ЦНС, почек, абдоминальная боль.
Сложность дифференциальной диагностики и ведения беременной с ТМА демонстрирует следующее клиническое наблюдение.
Пациентка, 34 года, доставлена в клинику по каналу СМП с диагнозом «Беременность 29–30 нед. Головное предлежание. Транзиторная ишемическая атака».
При поступлении пациентка предъявляла жалобы на слабость, незначительное головокружение, чувство онемения в правой руке. При сборе анамнеза нами было отмечено наличие хронического гастродуоденита, хронического цистита, хронического тонзиллита и тонзиллэктомии.
Из анамнеза: ветряная оспа, хронический цистит с 2013 г., хронический гастродуоденит (ремиссия). Аллергоанамнез не отягощен, наследственность по аутоиммунной патологии не отягощена, семейный тромботический анамнез не отягощен. В 1988 г. перенесла тонзиллэктомию без осложнений. Менструальная функция – без особенностей. Из гинекологических заболеваний: в 2007 г. проведена диатермокоагуляция шейки матки по поводу эрозии шейки матки, с 2011 г. – миома матки небольших размеров. Половая жизнь с 18 лет, брак 1 с 2008 г. Данная беременность у пациентки вторая. В 2013 г. самостоятельно наступила первая беременность, которая протекала без осложнений и завершилась своевременными родами – девочка весом 3130 г, ростом 53 см. Послеродовый период протекал без осложнений.
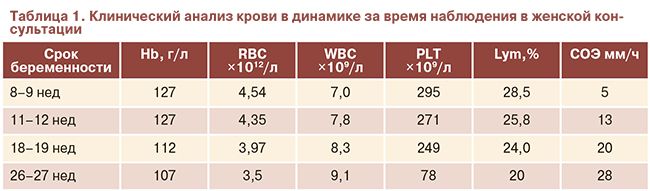
Настоящая беременность вторая, наступила самостоятельно, протекала с угрозой прерывания беременности в I триместре, по поводу чего проводились стационарное лечение, магнезиальная терапия, лечение прогестинами до 20 нед (Утрожестан 600 мг/сут вагинально). На сроке 26 нед при проведении глюкозотолерантного теста установлен диагноз гестационного сахарного диабета (глюкоза: 4,6–8,8–10,2 ммоль/л), который корригирован диетотерапией (cм. табл. 1).
За время наблюдения в женской консультации анализы мочи – без особенностей, мазок на онкоцитологию с поверхности шейки матки – без признаков атипии, серологические маркеры ИППП – отрицательные, консультация терапевта в сроке беременности 11–12 нед – без патологии, консультация офтальмолога – ангиопатия сетчатки, консультация стоматолога – полость рта санирована. По данным УЗИ в сроки 18–19 и 25 нед – размеры плода соответствуют сроку беременности, неполное предлежание плаценты, миома матки. При сборе анамнеза установлено, что за 2 нед до поступления в ГКБ им. С.С. Юдина в семье муж и ребенок перенесли вирусное заболевание с повышением температуры до 38°С и явлениями гастроэнтерита, пациентка самостоятельно применяла виферон в ректальных свечах. В течение около 1,5 недель отмечала пожелтение кожных покровов (на фоне лабораторного снижения числа тромбоцитов), нарастание слабости. Обращалась в женскую консультацию, была направлена к гематологу, однако на прием не попала. Сама пациентка за день до поступления отмечала жидкий стул в течение дня, боли в животе. Поводом для вызова СМП послужили головокружение и появление чувства онемения в правой руке. При поступлении состояние пациентки расценено как среднетяжелое. Кожный покров бледно-желтушного цвета, отмечается иктеричность склер. АД 120/70 мм рт. ст., пульс – 78 уд./мин, ритмичный. Частота дыхания 14 в мин, одышки нет. Живот увеличен за счет беременной матки, мягкий безболезненный. Моча желтого цвета. Матка в нормотонусе, не возбудима при пальпации. Проведено УЗИ – плод по показателям фетометрии соответствовал сроку гестации 29–30 нед, предполагаемый вес 1300 г. При допплерометрическом исследовании нарушений показателей кровотока в системе «мать–плацента–плод» не обнаружено.
Проведено лабораторное исследование. В клиническом анализе крови отмечено снижение уровня гемоглобина до 78 г/л, тромбоцитов до 9×109/л, уровень лейкоцитов – 7,4×109/л. Анализ мочи выявил высокий уровень протеинурии – 5,0 г/л. При биохимическом исследовании отмечено повышение уровня ЛДГ в 5 раз до (1134 г/л), повышение общего билирубина в 4,2 раза (71,6 г/л) преимущественно за счет непрямого (64,2 г/л) билирубина. Показатели ферментов печени – аспартат- и аланинаминотрансферазы, а также креатинин и мочевина – не выходили за пределы нормативных значений. Проведено УЗИ органов брюшной полости – выявлены диффузные изменения печени. УЗИ забрюшинного пространства и почек – без патологических изменений. МРТ головного мозга – патологических изменений не обнаружено. Выполнена катетеризация правой внутренней яремной вены.
По результатам лабораторных, клинических и инструментальных методов исследования выставлен диагноз: Беременность 29–30 нед. Головное предлежание плода. Низкая плацентация. Тромботическая микроангиопатия. Преэклампсия тяжелой степени. HELLP-синдром? Гемолитико-уремический синдром? Атипичный гемолитико-уремический синдром? Тромботическая тромбоцитопеническая пурпура? Острый жировой гепатоз?
Клиническими проявлениями ТМА стали:
- гемолитическая анемия (снижение уровня гемоглобина до 78 г/л, повышение уровня ЛДГ в 5 раз до 1134 г/л, повышение билирубина за счет непрямого);
- тромбоцитопения – снижение уровня тромбоцитов до 9×109/л,
- головокружение, слабость, онемение правой руки – как проявление ишемического поражения головного мозга.
Учитывая жизнеугрожающее состояние пациентки, отрицательную динамику лабораторных показателей (снижение уровня тромбоцитов до 9 тыс., высокие цифры ЛДГ, билирубина), консилиум принял решение о необходимости досрочного родоразрешения путем операции кесарева сечения. После предварительной подготовки в виде переливания тромбомассы и свежезамороженной плазмы проведена операция кесарева сечения. Родилась живая недоношенная девочка весом 1300 г, ростом 40 см, с оценкой состояния по шкале Апгар 5/7 баллов. Операция прошла без осложнений. Кровопотеря во время операции составила 700 мл. В последующем: рентгенологически – патологии не выявлено, УЗДГ вен нижних конечностей – признаков тромбоза не было, УЗИ органов малого таза – картина соответствовала срокам послеоперационного периода. В процессе дифференциальной диагностики рассматривалось предположение об акушерском ГУС. Однако отсутствие признаков почечной недостаточности, азотемии, отсутствие эндемического очага не позволяли думать о типичном ГУС. Атипичный ГУС (генетический дефект в системе комплемента) ставился под сомнение в связи с отсутствием наследственного анамнеза, неосложненным течением первой беременности. При наличии наследственно обусловленного дефекта комплемента первым его проявлением должна была быть первая беременность. Исследованиями последних лет установлено, что ассоциированный с беременностью аГУС составляет около 20% в структуре аГУС. Развивающийся в это время аГУС характеризуется агрессивным течением и неблагоприятным прогнозом. Мы не смогли выполнить больной генетические исследования, однако даже при наличии такой возможности у 30–50% пациентов не удается идентифицировать мутации в генах регуляторных белков комплемента, ведущие к развитию аГУС, и для установки диагноза таким образом достаточно лишь характерной клинико-лабораторной картины, как это имело место у нашей пациентки. Возможно, причиной развития такого тяжелого варианта ТМА стала беременность как таковая, а триггером могла послужить вирусная инфекция.
Таким образом, в качестве основных рассматривались два патологических состояния – тяжелая ПЭ и HELLP-синдром, а также ТТП. В пользу тяжелой ПЭ и HELLP-синдрома говорят высокий уровень протеинурии (5 г/л), наличие гемолиза, тромбоцитопении. Характерным признаком ТТП является резкое снижение уровня тромбоцитов и симптомы поражения ЦНС. В дифференциации этих патологических состояний существенную помощь оказывает определение уровня ADAMTS-13 в плазме. Если уровень ADAMTS-13 составляет <5%, это говорит в пользу ТТП. Сниженный уровень ADAMTS-13 >5%, но менее 30% преимущественно характерен для вторичной ТМА – тяжелых ПЭ и HELLP-синдрома. Нами предпринята попытка оценки уровня ADAMTS-13 в плазме. Однако забор образца был проведен после инфузии нескольких доз свежезамороженной плазмы. В нашем исследовании уровень ADAMTS-13 составил менее 5%. Однако анализ был проведен после начатой инфузии тромбомассы, кристаллоидов, плазмоинфузии, что не позволяет его рассматривать как достоверный.
Патогенетически обоснованной терапией при HELLP-синдроме и ТТП является переливание свежезамороженной плазмы и плазмаферез. Дальнейшее лечение после родоразрешения осуществлялось в отделении реанимации и интенсивной терапии. Проведены высокообъемная плазмоинфузия, плазмаферез, гемодиафильтрация. В анализах крови: тромобоциты 45–20–76–26×109/л; Hb 76–64 г/л; лейкоциты 11,6–7,4–14,0–19,4×109/л. В биохимических анализах крови: глюкоза – 5,2-7,7 ммоль/л; мочевина 4,3–14,1 ммоль/л, креатинин 73,9–122,7 мкмоль/л, общий белок 62-51 г/л, билирубин общий 71,6–30,4–185 мкмоль/л, АЛТ – 38–81 Ед/л, АСТ – 68–222 Ед/л, ЛДГ – 1134–3168 Ед/л.
В анализах мочи: кетоновые тела 15–0,9; билирубина нет; глюкоза 0–6,0; белок 5,0–0,5–0,3 г/л. Проводилось переливание эритроцитарной взвеси (всего 1680 мл), тромбоцитов (всего 710 мл). Проводился дискретный плазмаферез (плазма фильтрованная 5560 мл).
В течение пребывания пациентки в стационаре АД было в пределах 120/75–130/70 мм рт.ст.
Однако на фоне проведения указанной терапии состояние пациентки стало прогрессивно ухудшаться – присоединились явления печеночной недостаточности; появились симптомы энцефалопатии: уровень сознания – сомноленция, двигательный ответ – неразборчивая речь; отмечалось нарастание гипербилирубинемии до 134 г/л, уровня ЛДГ до 1500 г/л.
Несмотря на проводимую гемодиафильтрацию, отмечено нарастание печеночной недостаточности, уровня гипербилирубинемии, что было расценено как прогрессирование гемолиза в рамках установленного HELLP-синдрома. К сожалению, на 6-е сутки на фоне интенсивной реанимационной терапии вследствие прогрессирования печеночной недостаточности, отека головного мозга, дыхательной недостаточности констатирована смерть пациентки. Проведенное патологоанатомическое исследование позволило установить причину смерти – ПЭ тяжелой степени с развитием HELLP-синдрома, приведшего к ДВС-синдрому и полиорганной недостаточности. На секции выявлены признаки тяжелой преэклмапсии: ДВС-синдром с развитием множественных кровоизлияний во внутренних органах, слизистых и серозных оболочках, центролобулярные некрозы в печени; «шоковые» почки; анасарка. Гистологическое исследование плаценты: хроническая плацентарная недостаточность (плацентарная форма), гипоплазия плаценты (плодово-плацентарный коэффициент – 0,17 при норме 0,22–0,23).
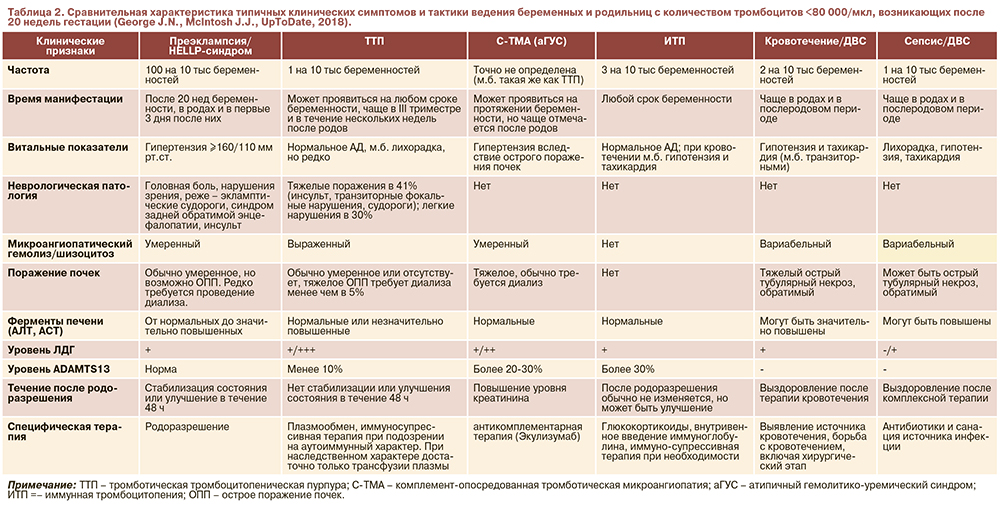
В заключение следует отметить, что тяжелая ПЭ и HELLP-синдром могут не иметь классической клинической картины, а проявляться в виде ТМА (тромбоцитопения, микроангиопатическая гемолитическая анемия) (см. табл. 2).
Выводы
Представленное наблюдение имеет целью познакомить практических врачей с особенностями течения, подходами к терапии и исходом атипичного ГУС, развившегося во время беременности и прогрессирующего после родоразрешения. Ассоциированная с беременностью и родами ТМА, независимо от срока ее развития, ставит перед врачом вопрос о необходимости разграничения различных микроангиопатических синдромов – в первую очередь тяжелой ПЭ и HELLP-синдрома, но также аГУС, ТТП и катастрофического АФС. Несмотря на сходство клинико-лабораторных проявлений этих видов патологии, подходы к их лечению и тактике ведения беременности различаются, в связи с чем своевременно и четко установленный диагноз необходимо рассматривать как основной фактор, определяющий прогноз и для матери, и для плода.


