
Что такое мезенхимальные дисплазии и каковы особенности их проявления у беременных?
Мезенхимальные дисплазии представляют собой группу заболеваний, характеризующихся врожденным дефектом соединительной ткани. Проявления заболеваний различны и включают в себя аномалии скелета, изменения со стороны органа зрения, кожи и подкожной клетчатки и многих других систем. Однако наибольшую опасность у таких больных представляет собой нарушение структуры соединительно-тканного компонента стенок сосудов различного калибра, так как именно с этим связаны тяжелые геморрагические осложнения при данных заболеваниях. Особенно высока вероятность развития таких осложнений во время беременности и в родах в связи с гемодинамическими и гормональными эффектами на стенки патологически измененных сосудов. Воздействия на стенку сосудов могут приводить к их дилатации, расслоению, разрывам, увеличению в размерах или формированию новых патологических сосудистых анастомозов (мальформаций). Характерной особенностью данной группы больных в мире является разрозненность и отсутствие их концентрации в единых медицинских центрах. Поэтому в современной литературе имеются лишь описания отдельных случаев беременности у таких пациенток. Большую проблему представляют собой больные с так называемыми «стертыми» формами мезенхимальных дисплазий, когда имеется недостаточно симптомов для установления точного диагноза. У таких больных заболевание часто протекает субклинически и вовремя не диагностируется. Риск возможных осложнений во время беременности, родов и послеродового периода недооценивается, что может приводить к летальным исходам.
Синдром Марфана: патогенез, клинические проявления, диагностика, особенности течения заболевания у беременных
Синдром Марфана представляет собой аутосомно-доминантное заболевание соединительной ткани, связанное с мутацией в гене фибриллина—одного из основных эластических компонентов соединительной ткани. Распространенность синдрома Марфана составляет 1 на 5-10 тыс. населения. Семейный анамнез заболевания прослеживается в 65-75% случаев, остальные случаи являются результатом вновь возникших мутаций.
Какова клиническая картина синдрома Марфана?
Наиболее частыми клиническими признаками заболевания являются прогрессирующая дилатация аорты, пролапс и недостаточность митрального и аортального клапанов, гипермобильность суставов, высокий рост, длинные конечности, арахнодактилия, деформация грудной клетки, сколиоз, подвывих хрусталика, миопия, готическое небо, стрии на коже. При рентгенологическом исследовании часто обнаруживаются эктазия твердой мозговой оболочки и протрузия вертлужных впадин. В некоторых случаях в анамнезе отмечаются повторные эпизоды пневмоторакса. Особое значение имеют проявления со стороны сердечно-сосудистой системы, так как именно с ними связана высокая летальность при этом заболевании.
На основании чего устанавливается диагноз синдрома Марфана? Каковы основные принципы клинической и молекулярной диагностики?
Фибриллин представляет собой гликопротеин с молекулярной массой 350 kD, синтезирующийся в виде предшественника с массой 375 kD и секретирующийся в интрацеллюлярный матрикс. Синтез фибриллина 1 типа кодируется геном, расположенным в хромосоме 15q21.1. Молекулярная диагностика синдрома Марфана затруднена, так как каждая мутация в гене фибриллина 1 уникальна и вероятность ее повторения чрезвычайно мала. Кроме того, мутации в гене фибриллина 1 обнаруживаются не только при синдроме Марфана, но и при сходных с ним заболеваниях, не имеющих тяжелых проявлений со стороны сердечно-сосудистой системы, например, при МАSS синдроме (myopia, mitral valve prolapse, aortic dilatation, skin involvement, skeletal involvement), изолированном подвывихе хрусталика.
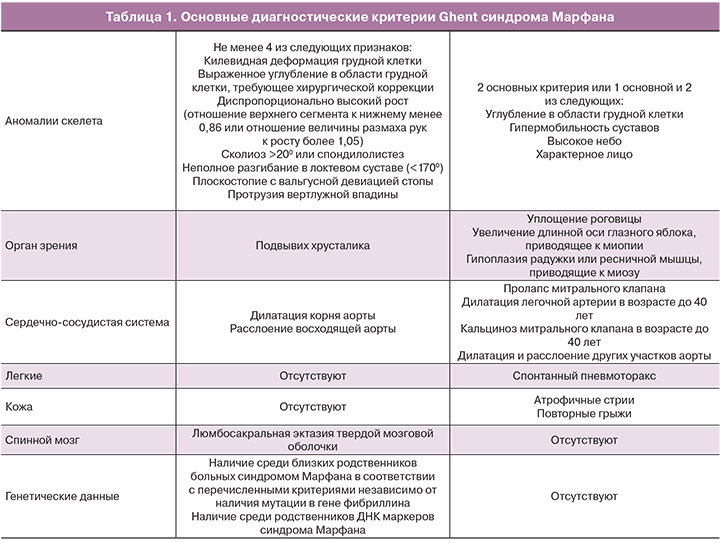
В настоящее время диагноз синдрома Марфана устанавливают в соответствии с диагностическими критериями Ghent (табл. 1).
Для постановки диагноза необходимо по 1 основному критерию со стороны 2 различных систем органов (4 признака со стороны скелета составляют 1 основной критерий) и 1 дополнительный критерий со стороны третьей.
Особо следует выделить так называемый «марфаноидный фенотип». Он включает в себя неярко выраженные признаки синдрома, такие как высокий рост, арахнодактилия, гипермобильность суставов, высокое готическое небо и другие симптомы, когда недостаточно критериев для постановки диагноза. Такие пациенты нуждаются в постоянном наблюдении, так как они имеют высокий риск развития более серьезных проявлений синдрома Марфана.
Каковы особенности течения беременности у пациенток с синдромом Марфана?
При ведении беременных с синдромом Марфана следует учитывать два основных обстоятельства [1, 2]:
Беременные с синдромом Марфана имеют высокий риск развития летальных осложнений со стороны сердечно-сосудистой системы (разрыва и расслоения аорты).
Заболевание наследуется у детей в 50% случаев.
Причинами высокого риска расслоения аорты во время беременности у пациенток с синдромом Марфана являются физиологическое увеличение объема циркулирующей крови и сердечного выброса на фоне врожденной аномалии коллагена. Определенную роль также имеют гормональные изменения. Гестационная гипертензия и преэклампсия резко увеличивают риск дилатации, расслоения и разрыва аорты [3, 4].
На основании чего можно диагностировать развитие расслоения аорты при синдроме Марфана?
Своевременная диагностика и терапия внезапного расслоения аорты жизненно необходимы, так как около 50% пациентов погибает в течение 48 часов после возникновения осложнения [5–7]. При беременности этот процент значительно выше. Основными симптомами расслоения аорты являются боль в груди, иррадиирующая в спину, плечи и живот. Наиболее опасными осложнениями являются экстравазальные кровотечения в перикард, плевральную полость, средостение, забрюшинное пространство, стенку легочной артерии, полости сердца. Кроме того, нередко наблюдаются симптомы, связанные с частичной или полной окклюзией различных артерий гематомой средней оболочки аорты. Окклюзия коронарных артерий может привести к внезапной смерти или инфаркту миокарда, общей сонной – к синкопальным состояниям, инсульту или коме, подключичной артерии – к ишемии верхних конечностей и парапарезам, межреберных или поясничных артерий – к ишемии спинного мозга. Иногда происходит окклюзия чревного ствола, почечной, мезентериальной или общей подвздошной артерий. Вследствие дилатации или расслоения аорты на уровне аортального клапана может развиться выраженная аортальная недостаточность и отек легких. Обструкция аорты или легочной артерии нередко приводит к циркуляторному коллапсу. При физическом обследовании часто выявляются дефицит пульса, диастолический шум на аорте, неврологические проявления (цереброваскулярные нарушения, потеря сознания, парапарезы или параплегии). При рентгенографии грудной клетки обнаруживается расширение средостения. Иногда имеются признаки гемоторакса (в основном левостороннего при расслоении дистального участка аорты). Однако данные рентгенографии неспецифичны и отсутствие патологических изменений на рентгенограммах не позволяет исключить диагноз. Золотым стандартом диагностики расслоения аорты является аортография. Однако во время беременности методами выбора в связи с их неинвазивностью и отсутствием отрицательного влияния на плод являются контрастная компьютерная томография, магнитно-резонансная томография (МРТ), трансэзофагеальная эхокардиография и ультразвуковое исследование. Дифференциальный диагноз расслоения аорты у беременных с синдромом Марфана проводится с такими острыми состояниями как эмболия околоплодными водами, инфаркт миокарда и аортальная регургитация, вызванные другими причинами, пневмоторакс, инсульт, разрыв матки, отслойка плаценты, тромбоз мезентериальных сосудов [8–10]. Как правило, диагноз расслоения аорты устанавливается post mortem.
Возможно ли снизить риск угрожающих жизни осложнений у беременных с синдромом Марфана?
Для предотвращения и своевременной коррекции угрожающих жизни осложнений на протяжении всей беременности больные с синдром Марфана должны находиться под тщательным наблюдением акушеров и сосудистых хирургов [11, 12]. Всем беременным с синдромом Марфана (даже ранее не имевшим признаков поражения сердечно-сосудистой системы) показано трансторакальное ультразвуковое исследование или МРТ в динамике [13, 14].
Во многих работах доказана эффективность β-адреноблокаторов для предотвращения прогрессирующей дилатации аорты [13, 14]. При расслоении дистального отдела аорты используется внутривенное введение β-адреноблокаторов до снижения частоты сердечных сокращений на 20%, снижения АД и сократимости левого желудочка. Хирургическое вмешательство при расслоении дистального отдела аорты показано при неэффективности медикаментозной терапии, разрыве или угрозе разрыва аорты, прогрессирующем расслоении аорты [14, 15]. При расслоении проксимального отдела аорты необходимо срочное оперативное вмешательство, так как только эта мера способна предотвратить летальный исход. Срочное хирургическое вмешательство показано также беременным с синдромом Марфана при увеличении диаметра аорты свыше 45 мм: на ранних сроках рекомендуется прерывание беременности, на поздних-кесарево сечение с последующей реконструктивной операцией на аорте [14, 15].
Другим частым показанием к неотложному оперативному вмешательству во время беременности у больных с синдромом Марфана является прогрессирующая аортальная недостаточность.
Каковы особенности родоразрешения пациенток с синдромом Марфана?
Оптимальным методом родоразрешения беременных с синдромом Марфана является кесарево сечение, что позволяет минимизировать гемодинамические изменения, связанные с вагинальным родоразрешением. Некоторые авторы рекомендуют одновременно с кесаревым сечением производить гистерэктомию, так как в послеродовом периоде у родильниц с синдромом Марфана часто отмечаются массивные маточные кровотечения. Причиной таких кровотечений является нарушение сократительной способности спиральных артерий, что имеет место и при других заболеваниях соединительной ткани, например, при синдроме Элерса–Данло.
Синдром Элерса–Данло: патогенез, клинические проявления, диагностика, особенности течения заболевания у беременных
Синдром Элерса–Данло представляет собой гетерогенную группу заболеваний соединительной ткани, характеризующихся гиперэластичностью кожи, гипермобильностью суставов, плохим заживлением ран с образованием неполноценных атрофичных рубцов и некоторыми другими симптомами. Распространенность этого синдрома варьирует от 1:560 000 до 1:5 000 [16].
На чем основана клиническая диагностика синдрома Элерса–Данло?
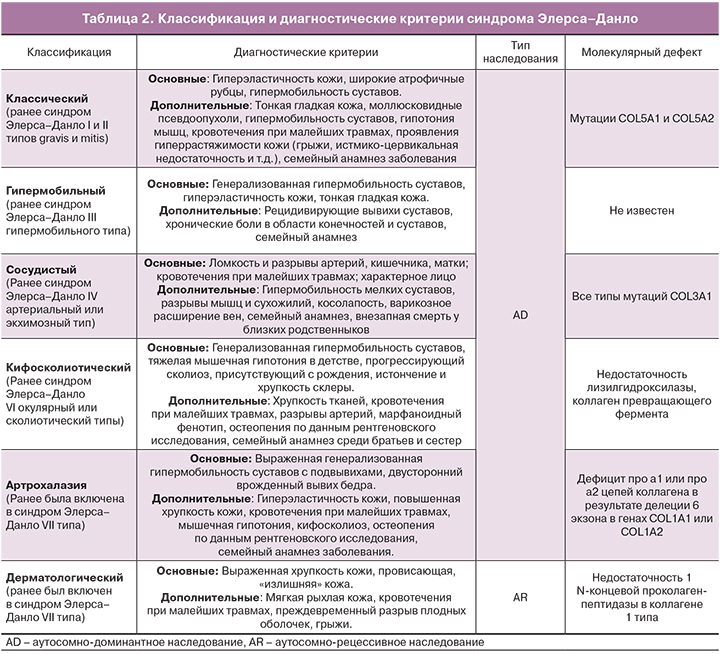
Первая классификация синдрома Элерса–Данло включала в себя 11 подгрупп. Впоследствии в 1986 году на Всемирном Конгрессе по наследственным заболеваниям соединительной ткани подтипы IX и XI были исключены из классификации. С развитием молекулярной биологии стало ясно, что подразделение на подгруппы лишь на основании клинических признаков не вполне корректно, поэтому была предложена новая классификация (1997 г.) (табл. 2). Именно на этой классификации в настоящее время основывается клиническая диагностика синдрома Элерса–Данло.
Кроме того, выделены отдельные редкие типы синдрома Элерса–Данло:
V тип: присутствуют основные клинические признаки классического типа, но менее выраженные. Наследуется по Х-сцепленному типу.
VIII тип (периодонтальный): Клинические признаки сходны с признаками классического типа плюс хрупкость десен. Наследуется аутосомно-доминантно.
X тип: Имеются основные признаки классического синдрома, выраженные в меньшей степени. Сопровождается нарушением агрегации тромбоцитов. Предполагается аутосомно-рецессивное наследование.
Из новой классификации исключены IX и XI типы.
N.B. Основные клинические критерии обладают высокой диагностической специфичностью, поскольку редко встречаются при других заболеваниях. Диагноз устанавливается на основании одного и более основных критериев. При наличии только дополнительных критериев диагноз сомнителен. По возможности желательно проведение молекулярной диагностики.
Каковы клинические проявления сосудистого типа синдрома Элерса–Данло?
Наибольшее значение в практике акушера-гинеколога имеет сосудистый тип синдрома Элерса–Данло, так как именно он сопровождается различными акушерскими и общемедицинскими проблемами, часто угрожающими жизни. При этом заболевании имеется дефект III типа коллагена, являющегося важнейшим компонентом сосудистой стенки и стенок желудочно-кишечного тракта. Повреждения артерий включают в себя разрывы, расслоения, артерио-венозные фистулы [16]. Разрывы артерий часто возникают без какой-то определенной причины. Наиболее опасные осложнения связаны с угрожающими кровотечениями, а также перфорацией кишечника. Так как хирургические вмешательства ограничены в связи с ломкостью тканей, эти кровотечения часто приводят к летальному исходу. Риск подобных осложнений возрастает с возрастом. В детстве их частота невелика, в то время как в возрасте до 20 лет они встречаются у 25% больных и более чем у 80% больных в возрасте до 40 лет. Даже незначительные травмы у пациентов с сосудистым типом синдрома Элерса–Данло приводят к образованию гематом, иногда довольно обширных. У таких пациентов часто образуются гематомы даже на месте манжетки при измерении артериального давления. Около 50% всех сосудистых повреждений составляют артерии груди и живота. Как правило, вовлекаются артерии среднего калибра. Часто кровотечения требуют неотложного хирургического вмешательства. Однако трудность заключается в том, что само хирургическое вмешательство может привести к массивным летальным кровотечениям из других артерий [16]. Вовлечение артерий верхних и нижних конечностей составляет около 25% среди различных сосудистых осложнений у пациентов с васкулярным типом синдрома Элерса–Данло. Разрывы артерий в анатомически закрытых пространствах приводят к образованию обширных гематом и часто заканчиваются летально. Предпочтительным является лигирование артерий, так как более сложные хирургические манипуляции могут привести к дальнейшим повреждениям.
Заподозрить сосудистый тип синдрома Элерса–Данло следует при наличии инсультов головного мозга в возрасте до 40 лет у пациентки или ее ближайших родственников. Одним из наиболее частых проявлений со стороны сосудов головного мозга у больных синдромом Элерса–Данло являются каротидно-кавернозные фистулы [16]. Частота внутричерепных кровоизлияний у больных с синдромом Элерса–Данло составляет около 4%. В половине случаев происходит разрыв ранее существовавших аневризм. Как можно более раннее установление диагноза у таких пациентов имеет первостепенную важность для выбора тактики дальнейшего обследования и хирургического вмешательства.
Поскольку коллаген III типа является одним из основных компонентов стенок желудочно-кишечного тракта, у пациентов с синдромом Элерса–Данло часто встречается перфорация кишечника. Летальность от перфорации кишечника у пациентов с синдромом Элерса–Данло относительно низка и составляет около 2%.
Дифференциальный диагноз сосудистого типа синдрома Элерса–Данло в первую очередь следует проводить с различными нарушениями системы гемостаза. Часто синдром является диагнозом исключения, то есть устанавливается после исключения всевозможных дефектов свертывающей системы крови. Кроме того, необходимо дифференцировать синдром Элерса-Данло с другими видами врожденных заболеваний соединительной ткани, в частности, с синдромом Марфана.
Молекулярная диагностика сосудистого типа синдрома Элерса–Данло основана на определении маркеров полиморфизма в локусе гена, расположенного в длинном плече 2 хромосомы (2q24.3-q31) и кодирующего III тип коллагена (COL3A1). Анализ этой последовательности позволяет идентифицировать генные аномалии и лежит в основе генетического консультирования и пренатальной диагностики заболевания. Однако данное исследование проводится лишь в ограниченном числе лабораторий.
В чем особенности ведения беременных с синдромом Элерса–Данло?
При планировании беременности у больных с синдромом Элерса–Данло следует учитывать подтип синдрома и присущие ему осложнения, а также риск передачи заболевания плоду (тип наследования). В связи с аутосомно-доминантным типом наследования риск передачи васкулярного типа синдрома Элерса–Данло плоду при наличии заболевания у одного из родителей составляет 50%, поэтому на этапе планирования беременности необходимо генетическое консультирование [16]. Беременность у пациенток с сосудистым вариантом синдрома Элерса–Данло связана с высоким риском разрыва артерий различного калибра, а, следовательно, массивных кровотечений, кровоизлияний и инсультов. Наиболее опасным в плане сосудистых осложнений является последний триместр беременности. Большую проблему представляет также внезапный разрыв матки. Материнская летальность при синдроме Элерса–Данло составляет по данным разных авторов от 10 до 25%. Риск осложнений также чрезвычайно высок во время родов и в раннем послеродовом периоде. У таких женщин часто наблюдаются массивные ранние послеродовые кровотечения, в основе которых лежит нарушение структуры спиральных артерий матки. Единственным способом остановки подобных кровотечений является гистерэктомия.
Для выявления передачи заболевания плоду используется генетический анализ мутации. Теоретически пренатальная диагностика возможна, однако в связи с высоким риском проведения самой процедуры для матери ее применение ограничено.
Вопрос о родоразрешении беременных с синдромом Элерса–Данло дискутабельный. С одной стороны, плановое кесарево сечение позволяет уменьшить риск осложнений, связанных с физической нагрузкой в родах (разрывы сосудов, инсульты) и маточными сокращениями (разрыв матки), а также обеспечить лучший контроль гемостаза. С другой – любое хирургическое вмешательство у таких больных сопряжено с высоким риском повреждения сосудов и массивного кровотечения, а также с осложнениями послеоперационного периода в связи с плохой заживляемостью ран и формированием неполноценного рубца. Все же по данным литературы большинство авторов отдают предпочтение операции кесарева сечения. Длительность послеоперационного наблюдения в этом случае должна быть увеличена в связи с возможными осложнениями.
Врожденная геморрагическая телеангиоэктазия (синдром Рендю–Ослера–Вебера): патогенез, клинические проявления, диагностика, особенности течения заболевания у беременных Врожденная геморрагическая телеангиоэктазия (HHT) или синдром Рендю–Ослера–Вебера), представляет собой группу аутосомно-доминантных заболеваний. Распространенность заболевания по различным данным составляет от 5 до 8 на 1000 населения [17].
Выделяют 2 формы заболевания (HHT 1 и HHT 2) в зависимости от мутации в гене эндолгина (9-я хромосома) или ALK-1 (12-я хромосома). Эти гены отвечают за синтез рецепторов к трансформирующему фактору роста и активину на эндотелии сосудов. При недостатке или изменении структуры этих рецепторов происходит нарушение формирования эластического и гладкомышечного компонентов сосудистой стенки.
Каковы основные клинические проявления синдрома?
Большую опасность при врожденной геморрагической телеангиоэктазии представляют артериовенозные мальформации в легких (PAVM-pulmonary arteriovenous malformations). Это прямые анастомозы между легочным артериальным и венозным руслом, минуя капилляры.
Степень выраженности клинических проявлений зависит от величины шунта между легочной артерией и веной. Основными методами диагностики PAVM являются рентгенологическое исследование грудной клетки, методика со 100% кислородом, контрастная эхокардиография, радионуклидное исследование, компьютерная томография, МРТ, ангиография легочной артерии [18, 19]. В связи с тем, что во время беременности у больных наследственной геморрагической телеангиоэктазией резко увеличивается рост легочных мальформаций, а также риск возникновения связанных с этим осложнений, скрининг в данной группе больных является обязательным. При отсутствии лечения летальность у больных с наследственной геморрагической телеангиоэктазией и легочными мальформациями составляет 50% по сравнению с 3% у больных, получивших какую-либо терапию. Основным методом лечения является чрезкожная эмболизация под контролем ангиографии легочной артерии [19].
У части больных с синдромом Рендю–Ослера обнаруживаются дефекты в системе гемостаза – нарушение функции тромбоцитов и системы фибринолиза. Примерно у 50% больных имеется нерезко выраженный синдром диссеминированного внутрисосудистого свертывания, редко переходящий в фульминантную форму.
На чем основана диагностика врожденной геморрагической телеангиоэктазии?
Для стандартизации подходов к диагностике и для предотвращения случаев гипердиагностики были выработаны следующие диагностические критерии врожденной геморрагической телеангиоэктазии по Curaçao.
- Спонтанные рецидивирующие носовые кровотечения
- Телеангиоэктазия в характерных местах:
- Губы.
- Слизистая рта.
- Пальцы.
- Слизистая носа.
- Висцеральные проявления:
- Телеангиоэктазии на слизистой желудочно-кишечного тракта (при наличии или отсутствии кровотечений)
- Артерио-венозные мальформации в легких.
- Артерио-венозные мальформации в печени.
- Артерио-венозные мальформации в головном мозге.
- Артерио-венозные мальформации в спинном мозге.
- Наличие среди родственников первой степени родства данного заболевания (в соответствии с перечисленными выше критериями).
- Диагноз врожденной геморрагической телеангиоэктазии является:
- Достоверным, если имеется, по крайней мере, 3 критерия.
- Вероятным или возможным, если имеется 2 критерия.
- Сомнительным, если имеется один критерий.
Все потомство больных наследственной геморрагической телеангиоэктазией имеет потенциальный риск манифестации заболевания в более позднем возрасте. При постановке диагноза следует исключить дефекты гемостаза. При наличии висцеральных проявлений заболевания у детей следует тщательно проверить остальных членов семьи. В будущем, вероятнее всего, клинический диагноз в соответствии с выше перечисленными критериями будет заменен молекулярными тестами, которые станут общедоступными.
В чем заключаются основные риски при ведении беременности у пациенток с синдромом Рендю–Ослера?
Беременность у пациенток с синдромом Рендю–Ослера связана с чрезвычайно высоким риском осложнений. Вопрос о беременности должен решаться индивидуально в каждом конкретном случае, однако благоприятный исход беременности следует ожидать лишь при правильном ее ведении, включающем своевременную диагностику и терапию осложнений заболевания. Имеются многочисленные данные о том, что во время беременности значительно усиливаются носовые кровотечения, телангиоэктазии на коже и слизистых становится более выраженными. Однако наибольшую опасность представляют осложнения, связанные с наличием легочных артериовенозных мальформаций, в частности массивные легочные кровотечения [19, 20].
Изменение гемодинамики и гормонального статуса беременных приводят к ухудшению состояния ранее существовавших мальформаций и росту новых. Чаще подобные осложнения имеют место во 2–3-х триместрах беременности, когда увеличение объема циркулирующей крови и сердечного выброса достигают максимума. Кроме того, высокий уровень прогестерона во время беременности способствует ослаблению стенок венозных сосудов, что также приводит к росту артериовенозных мальформаций. Вопрос о проведении эмболизации легочных мальформаций у беременных, страдающих наследственной геморрагической телеангиоэктазией является дискутабельным [19, 20].
Какой метод родоразрешения показан пациенткам с синдромом Рендю–Ослера?
На данный момент не существует четких рекомендаций о наиболее целесообразном методе родоразрешения беременных с синдромом Рендю–Ослера. Отсутствуют исследования о сравнительной безопасности консервативного и оперативного родоразрешения. В большинстве наблюдений различных авторов беременность у женщин с синдромом Рендю–Ослера завершилась плановым или экстренным кесаревым сечением. Плановое оперативное родоразрешение, по-видимому, показано беременным с имеющимися легочными или церебральными мальформациями (или подозрением на их наличие). В связи с возможными артерио-венозными мальформациями спинного мозга таким беременным не рекомендуется проведение эпидуральной анестезии.
Таким образом, наиболее безопасной стратегией для женщин с наследственной геморрагической телеангиоэктазией является планирование беременности с тщательным обследованием до беременности, в частности на предмет наличия артериовенозных мальформаций в легких и головном мозге (ангиография, МРТ, компьютерная томография) и при необходимости их хирургической коррекцией. Кроме того, следует учитывать, что риск наследования заболевания ребенком составляет 50%.
Заключение
Беременность у больных с мезенхимальными дисплазиями сопряжена с высоким риском осложнений со стороны матери и плода, в связи с чем для успешного исхода беременности у таких больных следует рекомендовать мультидисциплинарный подход, а также тщательное динамическое наблюдение с широким спектром лабораторно-инструментальных исследований, начиная с момента планирования беременности и заканчивая послеродовым периодом. Ввиду высокого риска угрожающих жизни осложнений при родоразрешении через естественные родовые пути таким пациенткам показано родоразрешение путем операции кесарева сечения. В связи высокой частотой осложнений послеродового периода у больных с мезенхимальными дисплазиями целесообразным является увеличение длительности наблюдения за такими пациентками в послеродовом периоде. Все новорожденные от матерей с мезенхимальными дисплазиями должны быть тщательно обследованы в связи с высокой вероятностью наследования заболевания ребенком. Беременные со стертыми формами заболеваний (марфаноидный фенотип, пролапс митрального клапана III степени, миопия высокой степени или подвывих хрусталика, гипермобильность суставов, гиперэластичность кожи, многочисленные атрофичные рубцы, телеангиоэктазии на коже и слизистых, склонность к частым носовым кровотечениям и другие симптомы), когда недостаточно критериев для установления диагноза, согласно современным критериям диагностики, составляют группу повышенного риска и требуют такого же подхода, как и беременные с достоверным диагнозом мезенхимальных дисплазий.


